

Post-traumatic stress disorder (PTSD) is now well established as a discrete psychiatric disorder with evidence of a distinct neurobiology. Its features have been refined since it was first introduced as a diagnosis in DSM–III (American Psychiatric Association, 1980) and the past decade has seen an exponential rise in the number of publications relating to it. The disorder is characterised by re-experiencing (e.g. nightmares and flashbacks), avoidance, numbing of general responsiveness, and hyperarousal (e.g. irritability and hypervigilance) following a traumatic event. It has been estimated to have a lifetime prevalence of 8% in the USA, is deemed to be chronic if it lasts for more than 3 months and runs a course whereby around a half recover within 2 years but about a third continue to have the diagnosis 6 years later (Reference Kessler, Sonnega and BrometKessler et al, 1995).
Factors associated with the development of PTSD include lack of perceived social support after the event, marked initial distress, high-impact trauma (rape has consistently been associated with the highest rates), dissociation at the time, past psychiatric history and female gender (Reference Brewin, Andrews and ValentineBrewin et al, 2000; Reference Ozer, Best and LipseyOzer et al, 2003). Over half of PTSD sufferers will also have another psychiatric disorder (Reference Kessler, Sonnega and BrometKessler et al, 1995). As a diagnosis, PTSD has always been associated with a degree of controversy. Some have criticised the very existence of PTSD, arguing that it is a Western social construct; others acknowledge its existence but are concerned by probable over-diagnosis (e.g. Reference TyrerTyrer, 2005).
Aside from the controversy over the diagnosis of PTSD, its management has also been controversial. Interest in the prevention of its development following a traumatic event provoked attempts to discover effective early psychological interventions. One of these, psychological debriefing, has now been shown to lack evidence of efficacy (Rose et al, 2005) but trauma-focused cognitive–behavioural interventions for symptomatic individuals have been shown to be efficacious (e.g. Reference Bryant, Harvey and DangBryant et al, 1998; Reference Ehlers, Clark and HackmannEhlers et al, 2003; Reference Bisson, Shepherd and JoyBisson et al, 2004).
Of more pertinence here is the possibility of a new area of controversy in the management of PTSD, that of the effectiveness or otherwise of pharmacological approaches. Clinical practice guidelines commissioned by the National Institute for Health and Clinical Excellence (NICE) recommend that medication should be considered a second-line treatment for PTSD, behind trauma-focused psychological treatments such as trauma-focused cognitive–behavioural therapy and eye movement desensitisation and reprocessing (National Collaborating Centre for Mental Health, 2005). My primary aim in this article is to critically review the evidence for a pharmacological approach to the management of PTSD, to allow readers to understand the evidence on which the NICE guidelines have been based and draw their own conclusions regarding the role of medication in PTSD.
The neurobiology of PTSD
Our understanding of the neurobiology of PTSD has been greatly informed by work on memory and fear conditioning. The hippocampus and other areas of the temporal lobe are believed to mediate conscious memories, for example the exact details of a traumatic event. The amygdala is believed to be important in the mediation of unconscious memories, for example the autonomic aspects of the classic fear response. The amygdala receives information about external stimuli and is involved in determining their significance. This then triggers emotional responses, including ‘fight, flight or freezing’ and alterations in stress hormones and catecholamines.
Connections between the amygdala, hippocampus and medial prefrontal cortex have been implicated in determining the final fear response. Hippocampal lesions have been associated with a stronger fear response and smaller hippocampal volume has been associated with PTSD in several studies, probably representing a pre-existing vulnerability to its development rather than a neurotoxic consequence (Reference Gilbertson, Shenton and CiszewskiGilbertson et al, 2002). Neuroimaging studies of people with PTSD have shown decreased activity in medial prefrontal and anterior cingulate areas to be correlated with increased activity in the amygdala (Reference Bremner, Staib and KaloupekBremner et al, 1999; Reference Shin, Orr and CarsonShin et al, 2004). This has resulted in the proposal that PTSD represents a failure of medial prefrontal and/or anterior cingulate networks to regulate amygdala activity, resulting in hyperreactivity to threat (Reference BremnerBremner, 1999).
One of the most enduring neurophysiological theories in recent times has been that of enhanced negative feedback in the hypothalamic–pituitary–adrenal axis. Several studies have found low cortisol levels in people with PTSD and an opposite response to the dexamethasone suppression test than that seen with severe depression, i.e. there is over-suppression of cortisol release. However, more recent studies have not consistently supported this finding (Reference Young and BreslauYoung & Breslau, 2005). The finding of increased plasma catecholamine levels in PTSD sufferers has also evoked considerable interest, including the suggestion that an initial adrenergic (adrenaline and noradrenaline) surge may be associated with the laying down of traumatic memories (Reference PitmanPitman, 1989).
Our improved understanding of the neurobiology of PTSD does not appear to have driven most studies of the efficacy of pharmacological agents in the treatment of the disorder, but it has led to the development of hypotheses for the potential effectiveness of some drugs (for example propranolol and hydrocortisone as early interventions, see below). It has also resulted in attempts to determine the impact of drugs on the neurobiological processes themselves. Citalopram, for example, has now been shown to influence the acquisition of fear conditioning (Reference Burghardt, Sullivan and McEwenBurghardt et al, 2004).
Determining the efficacy of interventions
Various levels of evidence can be used to assess the efficacy of interventions but replicated, multicentre, large-scale randomised controlled trials (RCTs) should be considered the gold standard. This level of evidence is lacking for most pharmacological approaches to PTSD treatment, with the notable exceptions of paroxetine and sertraline. Nevertheless, there have been RCTs (albeit not necessarily to the gold standard) of several pharmacological approaches and here I consider the results of those for drugs that are currently available.
The efficacy of the drugs is described in terms of reduction in clinician-assessed severity of PTSD symptoms using the results of the NICE meta-analysis in this area (National Collaborating Centre for Mental Health, 2005). In studies that did not include a clinician-rated outcome measure the results of the Impact of Event Scale, a self-report measure, are given. Box 1 outlines the statistical terms used in the presentation of the results.
Box 1 Presentation of results
-
• k = number of trials
-
• n = total number of participants included in the analysis
-
• s.m.d. (standardised mean difference): a statistical means of determining the effect size of a continuous outcome measure. A value of 0 represents no difference between the groups
-
• Effect size: in general it is accepted that effect sizes of < 0.5 are low, < 0.8 medium and > 0.8 large (Reference CohenCohen, 1988)
-
• 95% confidence intervals (95% CIs): these figures represent the range of standardised mean differences within which we can be 95% confident that the true level of difference falls
-
• RR (relative risk): in the prevention studies the RR is used instead of the s.m.d. to describe the efficacy of the drug in preventing the development of PTSD. This statistic compares the risk of developing PTSD for the active drug group with that for the placebo group. A relative risk of 1 represents no difference between the groups
Prevention of PTSD
Three small RCTs of early pharmacological interventions to prevent PTSD have been published, all of which are likely to have been underpowered to detect clinically important differences unless of a very large magnitude. The first was based on the cortisol findings described above and concerned the administration of intravenous hydrocortisone to victims of septic shock on an intensive care unit in Switzerland (Reference Schelling, Briegal and RoozendaalSchelling et al, 2001). The results were not statistically significant but those receiving hydrocortisone tended to be less likely to develop symptoms of PTSD (k = 1; n = 20; RR = 0.17; 95% CI 0.03–1.17). This has not been replicated in other populations, although a case study of four individuals with chronic PTSD suggests that it may be worthy of further investigation (Reference Aerni, Traber and HockAerni et al, 2004).
The second study concerned propranolol, a beta-blocker. It was based on the hypothesis that an adrenergic surge beginning almost immediately after a traumatic event is associated with the development of traumatic memories. Reference Pitman, Sanders and ZusmanPitman and colleagues (2002) hypothesised that, for propranolol to be successful, individuals would have to start taking it within 6 h of the trauma. The logistic implications of running a trial with this constraint are daunting, but all credit to the researchers in completing a small study of individuals who were randomly allocated to receive 40 mg propranolol or a placebo four times a day for 10 days. There was no significant difference in rates of PTSD between the two groups at 1 month (k = 1; n = 41; RR = 1.14; 95% CI 0.55–2.35) or 3 months (k = 1; n = 41; RR = 1.28; 95% CI 0.69–2.38), with the trend being in favour of the placebo group. However, the propranolol group became less physiologically aroused when they listened to an account of the traumatic event, which indicates the possibility of some effect but not enough to recommend routine prescribing.
Finally, there has been one small RCT of temazepam given shortly after a traumatic event (Reference Mellman, Bustamante and DavidMellman et al, 2002). This is of interest because it has been argued that benzodiazepines may hinder the processing of trauma, but on closer scrutiny such assertions appear to be based more on anecdote and, possibly, an anti-prescribing stance than evidence. From a mean of 14 days after the trauma, individuals in the treatment arm received 30 mg temazepam daily for 5 days, then 15 mg daily for 2 days. There was no statistical difference in rates of PTSD at the 6-week follow-up point, although a trend was found in favour of the placebo group (k = 1; n = 22; RR = 3.2; 95% CI 0.54–18.98). The temazepam group had slept significantly better on the first night, but there was no significant difference between groups at follow-up.
In summary, there is not enough evidence to advocate the routine prescription of medication to prevent PTSD. There is some evidence for intravenous hydrocortisone in people with septic shock, but clearly it is difficult to generalise these results to other populations. There is also some evidence that temazepam may help with acute insomnia following traumatic events. There has been no research to date exploring the potential of more commonly used agents such as antidepressants in the immediate or early aftermath of a traumatic event.
Treatment of PTSD
Much more research has been conducted regarding the efficacy of drugs in the treatment of established chronic PTSD (Fig. 1). Perhaps not surprisingly the selective serotonin reuptake inhibitors (SSRIs) have been investigated more than any other family of drugs, indeed more than any other intervention in the field. The number of individuals entered into RCTs of SSRIs outnumbers those included in trauma-focused cognitive–behavioural therapy trials by over 2:1. There are probably several reasons for this, including the willingness of the pharmaceutical industry to fund studies of drugs with the potential to gain another indication for prescribing. It is a sad fact that drugs that have shown some potential but have lost their patent are not investigated to the same degree.
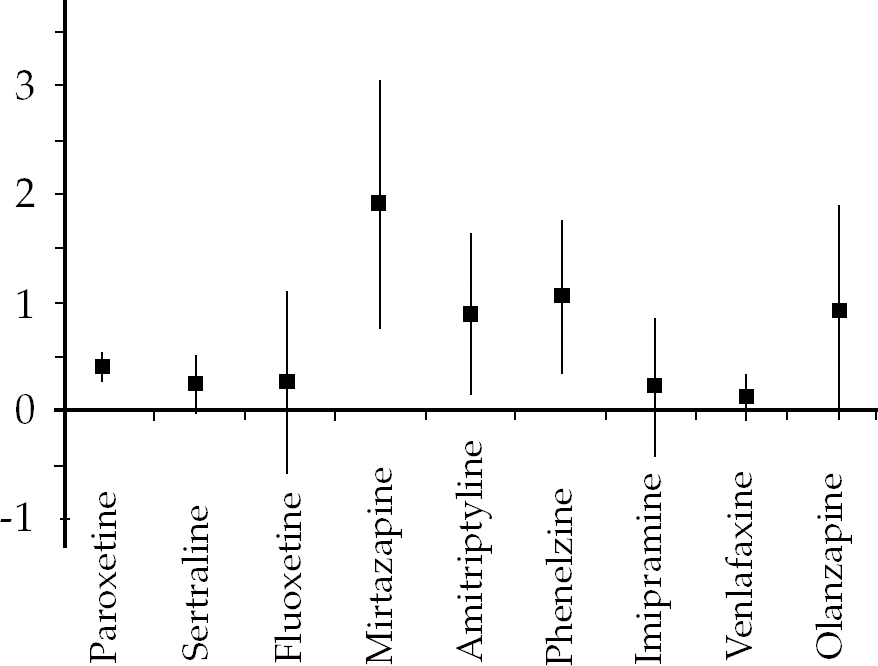
Fig. 1 Treatment of chronic PTSD: standardised mean differences with 95% confidence intervals.
Selective serotonin reuptake inhibitors
Paroxetine
There have been three main well-designed double-blind RCTs of paroxetine. Two of these have been published (Reference Marshall, Beebe and OldhamMarshall et al, 2001; Reference Tucker, Zaninelli and YehudaTucker et al, 2001), the other (SKB 627) has not but the results are within the public domain (National Collaborating Centre for Mental Health, 2005: p. 69). Paroxetine shows a statistically significant positive effect over placebo (k = 3; n = 1070; s.m.d. = −0.42; 95% CI −0.55 to −0.3). The tight CIs suggest that this is likely to be an accurate estimate of its efficacy, but the critical question is how clinically significant this effect is: enough for it to be granted a licence for PTSD by the UK authorities but not enough for it to be recommended in the NICE guidelines (National Collaborating Centre for Mental Health, 2005) as a first-line treatment! The Guideline Development Group set a priori limits on what they would consider clinically meaningful, and as a result gave only a limited recommendation to paroxetine as a second-line treatment. This, along with the widely publicised other potential problems associated with paroxetine (Reference DuffDuff, 2004), should result in its cautious use for PTSD.
Sertraline
One of the most interesting things about sertraline is that, although it is widely recommended as an effective treatment for PTSD (e.g. Reference Friedman, Davidson, Mellman, Foa, Keane and FreidmanFriedman et al, 2000; Reference Stein, Zungu-Dirwayi and Van der LindenStein et al, 2004), PTSD is an indication for its use in the UK in females but not in males. This suggests that the data presented to the authorities were not totally convincing. In fact, the NICE figures from four published (Reference Brady, Pearlstein and AsnisBrady et al, 2000; Reference Davidson, Rothbaum and van der KolkDavidson et al, 2001a , Reference Davidson, Baldwin and Stein2006; Reference Zohar, Amital and MiodownikZohar et al, 2002) and two unpublished studies (Pfizer 588; Pfizer 589) just fail to achieve statistical significance, with a trend in favour of sertraline (k = 6; n = 1123; s.m.d. = −0.26; 95% CI −0.51 to 0.00). Interestingly, the Guideline Development Group was aware of the two unpublished studies of sertraline held by Pfizer (Pfizer 588; Pfizer 589). Despite several requests the full results were not forthcoming, although enough information was obtained to include the studies in the final meta-analysis (National Collaborating Centre for Mental Health, 2005: p. 71). The inclusion of these two studies reduced the apparent efficacy of sertraline. Such experiences encourage close scrutiny of efficacy claims and beg the question ‘How many other unpublished trials are there?’. Calls for the pre-registration of RCTs and an undertaking to place all results in the public domain seem to be well founded.
Fluoxetine
Fewer individuals have participated in fluoxetine trials than in trials for the other two SSRIs, and only one trial (Reference Martenyi, Brown and ZhangMartenyi et al, 2002) used a standardised clinician assessment of the severity of PTSD symptoms as a primary outcome measure. The results are less convincing than those for paroxetine and do not reach statistical significance (k = 1; n = 301; s.m.d. = −0.28; 95% CI 0.54 to −1.2), but this may be explained by insufficient numbers to show a real but modest effect.
Tricyclics and monoamine oxidase inhibitors
The RCTs of tricyclic antidepressants and monoamine oxidase inhibitors (MAOIs) are older than those of SSRIs and their quality is inferior. It is disappointing that more trials have not been carried out, particularly given the encouraging results of those that have been published. None of the trials used a clinician-rated outcome measure and therefore the results of the Impact of Event Scale, a self-report measure, are given.
Amitriptyline
The one study (Reference Davidson, Kudler and SmithDavidson et al, 1990) that considered the efficacy of amitriptyline was positive but with very large confidence intervals, meaning that its true effect could be anything between low and very large (k = 1; n = 33; s.m.d. = −0.9; 95% CI −1.62 to −0.18).
Imipramine
The evidence for imipramine (Reference Kosten, Frank and DanKosten et al, 1991) is weaker than that for amitriptyline and the effect does not achieve statistical significance, although the wide confidence intervals mean that its true effect could be a positive one (k = 1; n = 41; s.m.d. = −0.24; 95% CI −0.86 to 0.38).
Phenelzine
Phenelzine is the only available MAOI with an evidence base for the treatment of PTSD (Reference Kosten, Frank and DanKosten et al, 1991) and, like amitriptyline, although the true magnitude of its effect is not known it appears to be efficacious (k = 1; n = 37; s.m.d. = −1.08; 95% CI −1.75 to −0.36).
Other drugs
Mirtazapine
There has been one small RCT (Reference Davidson, Weisler and ButterfieldDavidson et al, 2003) of mirtazapine which was strongly in favour of the drug (k = 1; n = 21; s.m.d. = −1.89; 95% CI −3.00 to −0.78). It is important to be cautious when interpreting the true effect of any intervention on the basis of one small RCT, but these results suggest that mirtazapine is worthy of further investigation.
Venlafaxine
The results of the single relatively large RCT of venlafaxine (Reference Davidson, Baldwin and SteinDavidson et al, 2006) were disappointing and did not achieve statistical significance. They suggest that any positive effects are likely to be at best modest overall (k = 1; n = 358; s.m.d. = −0.14; 95% CI −0.35 to 0.06).
Olanzapine
The one small RCT of olanzapine (Reference Butterfield, Becker and ConnorButterfield et al, 2001) as a first-line treatment for PTSD was not positive (k = 1; n = 11; s.m.d. = 0.04; 95% CI −1.19 to 1.26). However, an underpowered RCT of olanzapine in the augmentation of SSRIs (Reference Stein, Kline and MatloffStein et al, 2002) showed a trend in favour of olanzapine (k = 1; n = 19; s.m.d. = −0.92; 95% CI −1.88 to 0.04), suggesting that further work in this area is warranted.
Risperidone
Risperidone for PTSD has been investigated only as an adjunct to other medications (Reference Hamner, Faldowski and UlmerHamner et al, 2003) and it did not appear to be efficacious (k = 1; n = 37; s.m.d. = 0.1; 95% CI −0.55 to 0.74).
Non-RCT trials
No other true RCTs were identified by the Guideline Development Group, although several other drugs have been used for PTSD. For example, some benzodiazepines, including clonazepam, alprazolam, carbamazepine, clonidine and prazosin, have been reported as being efficacious in open-label trials, case reports or case series. Further evaluation of these and other drugs will be required before it can be determined whether they are effective. However, the adage ‘absence of evidence is not evidence of absence of effect’ holds true.
Prescribing for PTSD
The disappointing evidence base for the efficacy of drugs in PTSD should lead to reduced and more cautious prescribing for this condition. The true role of drugs in the management of PTSD remains unclear. The NICE guidance (National Collaborating Centre for Mental Health, 2005) is that drugs should only be used as a second-line treatment, when trauma-focused psychological treatment has not worked, is contraindicated or not wanted by the patient, or to treat a comorbid condition. However, the only way to properly determine the comparative efficacy of drug treatment and trauma-focused psychological therapy is through adequate head-to-head studies, which are yet to be conducted. At present trauma-focused psychological treatments are not readily available in the National Health Service and there are often long waits for them. This is likely to mean that delay in access to such treatment will commonly result in the prescribing of medication for PTSD as a first-line intervention.
The NICE guidelines recommend paroxetine and mirtazapine as the drugs with most evidence for widespread use, with amitriptyline and phenelzine being reserved for initiation by secondary care professionals. There are limitations to all these drugs both in terms of limited evidence (see above) and their side-effect profiles. It seems likely that many prescribers will continue to feel more comfortable prescribing antidepressants they are more familiar with, despite the absence of an evidence base.
What dose?
If medication is prescribed it is difficult to determine the best dose from the RCTs because it is well recognised that RCTs that allow increases in dose tend to report high mean doses. More informative is the fixed-dose RCT of paroxetine (Reference Marshall, Beebe and OldhamMarshall et al, 2001), which showed that 20 mg had much the same effect as 40 mg. In practice it seems sensible to increase the dose to the minimum known to be possibly effective and then to monitor for a month before considering further increase, although some studies have shown increasing benefits beyond a month without an increase in dose. As with all drugs, the actual dose will also be influenced by side-effects.
How long should treatment continue?
If effective, the NICE guidelines recommend that treatment is continued for at least a year (National Collaborating Centre for Mental Health, 2005). There is no good evidence for this recommendation, which was largely based on clinical experience, although one trial of sertraline showed a lower relapse rate in those continuing on the active medication for 28 weeks after a positive response than those on placebo (Reference Davidson, Pearlstein and LondborgDavidson et al, 2001b ).
Adverse effects
The side-effects associated with the use of these drugs for other conditions appear to apply to their use in PTSD. The NICE guidelines give no information on adverse effects by study, although the Guideline Development Group found no significant difference between paroxetine and placebo on reducing the likelihood of leaving treatment early (k = 3; n = 1196; RR = 0.95; 95% CI 0.79–1.15). The same applies for sertraline (k = 6; n = 1148; RR = 1.10; 95% CI 0.90–1.33), mirtazapine (k = 1; n = 29; RR = 1.20; 95% CI 0.29–2.82) and venlafaxine (k = 1; n = 358; RR = 0.83; 95% CI 0.62–1.12), although fluoxetine was associated with a greater drop-out rate than placebo (k = 1; n = 131; RR = 0.51; 95% CI 0.28–0.96).
Some individual studies have reported adverse effects. One RCT (Reference Davidson, Rothbaum and van der KolkDavidson et al, 2001a ) found that, compared with placebo, sertraline significantly increased insomnia, diarrhoea and nausea, and decreased appetite. An RCT comparing paroxetine with placebo (Reference Tucker, Zaninelli and YehudaTucker et al, 2001) found that nausea, somnolence, dry mouth, asthenia and abnormal ejaculation had an incidence of at least 10% and twice that of placebo. In one RCT (Reference Davidson, Weisler and ButterfieldDavidson et al, 2003) three people taking mirtazapine withdrew because of adverse effects, including sedation, panic attacks, increased anxiety and irritability. Three people taking placebo withdrew because of pain, or lack of efficacy; more people taking mirtazapine had increased appetite and weight gain. It is also important to consider the possibility of a discontinuation syndrome with the SSRIs, and paroxetine in particular, and to follow the well-publicised prescribing details (Reference DuffDuff, 2004).
What if the patient fails to respond?
If an individual fails to respond they should be reassessed to confirm that PTSD is the correct diagnosis. If it is and trauma-focused psychological treatment has not been offered and is indicated attempts should be made to arrange this. As regards alternative pharmacological approaches, increase in the dose of a tolerated antidepressant would be appropriate and, if this is unsuccessful, change to an alternative antidepressant. If this does not help, augmentation with olanzapine should be considered. In clinical practice several individuals will remain symptomatic and their distress and/or the risks associated with their condition will be considered severe enough to try other pharmacological approaches, although none can be recommended given the current evidence base.
Comorbidity
It is vital to determine whether an individual with PTSD has a comorbid psychiatric disorder. Over half of sufferers will have and often this comorbid condition will need treating in its own right. The most common co-diagnoses are depressive disorders, other anxiety disorders and substance misuse. As a general rule it is important to try to reduce substance use first and then reassess before treating the PTSD, although some centres have reported success with a combined treatment approach. With depression and anxiety it often depends which appears to be the primary disorder. If the PTSD predominates then treatment of this first is appropriate and often associated with a concomitant reduction of depressive and anxiety symptoms (National Collaborating Centre for Mental Health, 2005). For some individuals treatment of their depression may help them to benefit more from psychological treatment for their PTSD.
The future
There is clearly an urgent need to identify more effective pharmacological approaches for the management of PTSD. Despite the more encouraging picture in terms of effectiveness of psychological treatments, not all PTSD sufferers will be able to engage with these and some would prefer a pharmacological alternative. It is to be hoped that in the future the development of novel agents will be based on an improved understanding of the neurobiology of PTSD and translational research, the collaborative interaction of laboratory and clinical medicine.
Declaration of interest
J. I. B. was Co-Chair of the Guideline Development Group and co-author of the NICE guidelines on PTSD (National Collaborating Centre for Mental Health, 2005). His department has received funding from Wyeth for participation in an RCT of venlafaxine in the treatment of PTSD.
MCQs
-
1 In the UK, PTSD is an indication for the following in men and/or women:
-
a sertraline
-
b fluoxetine
-
c mirtazapine
-
d imipramine
-
e amitriptyline.
-
-
2 The autonomic aspects of the classic fear response are believed to be mediated by the:
-
a frontal cortex
-
b cerebellum
-
c hippocampus
-
d amygdala
-
e parietal lobe.
-
-
3 RCTs have shown the following not to be statistically significantly superior to placebo for PTSD:
-
a paroxetine
-
b phenelzine
-
c mirtazapine
-
d venlafaxine
-
e amitriptyline.
-
-
4 The NICE guidelines for PTSD recommend mirtazapine or paroxetine in the following circumstances:
-
a first-line treatment of PTSD
-
b second-line treatment of PTSD
-
c for combined use
-
d for females only
-
e only to be initiated in secondary care.
-
-
5 As a treatment for PTSD there is no RCT evidence regarding the efficacy (or not) of:
-
a mirtazapine
-
b venlafaxine
-
c carbamazepine
-
d olanzapine
-
e fluoxetine.
-
MCQ answers

| 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a | T | a | F | a | F | a | F | a | F |
| b | F | b | F | b | F | b | T | b | F |
| c | F | c | F | c | F | c | F | c | T |
| d | F | d | T | d | T | d | F | d | F |
| e | F | e | F | e | F | e | F | e | F |


eLetters
No eLetters have been published for this article.