

This is the second of two articles reviewing the antipsychotic drugs often referred to as ‘first-generation’, ‘conventional’ or ‘typical’ – in reality, compounds marketed before 1993. The first article considered phenothiazines (Reference OwensOwens 2012, this issue). Here, I review the thioxanthenes, butyrophenones (plus the closely related diphenylbutylpiperidines) and substituted benzamides currently available in the UK in oral formulations. As maximising prescribing choices depends on an individualised ‘risk–benefit appraisal’, the article also considers safety and tolerability issues more prevalent or reported more frequently with the older compounds.
Thioxanthenes
Thioxanthenes were synthesised in Denmark in 1958 and represent only a slight structural modification to the phenothiazine molecule. However, the substitution of carbon for nitrogen at position 10 means that these molecules exhibit stereoisomerism – i.e. R2 substitutions can attach in mirror-image ways. The pharmacological consequence is substantial, in that only one isomer (the cis or Z)Footnote a is an effective D2 dopamine receptor antagonist. Although widely used internationally, the first thioxanthene, chlorprothixene, was not marketed in the UK.
Flupentixol (flupenthixol) (Table 1)
Flupentixol is the thioxanthene analogue of fluphenazine and, although mainly used in the UK in its decanoate formulation as a depot, is also available (as dihydrochloride) in tablet form, a presentation traditionally popular with general practitioners.
TABLE 1 Non-phenothiazine older antipsychotics for oral use: thioxanthenes
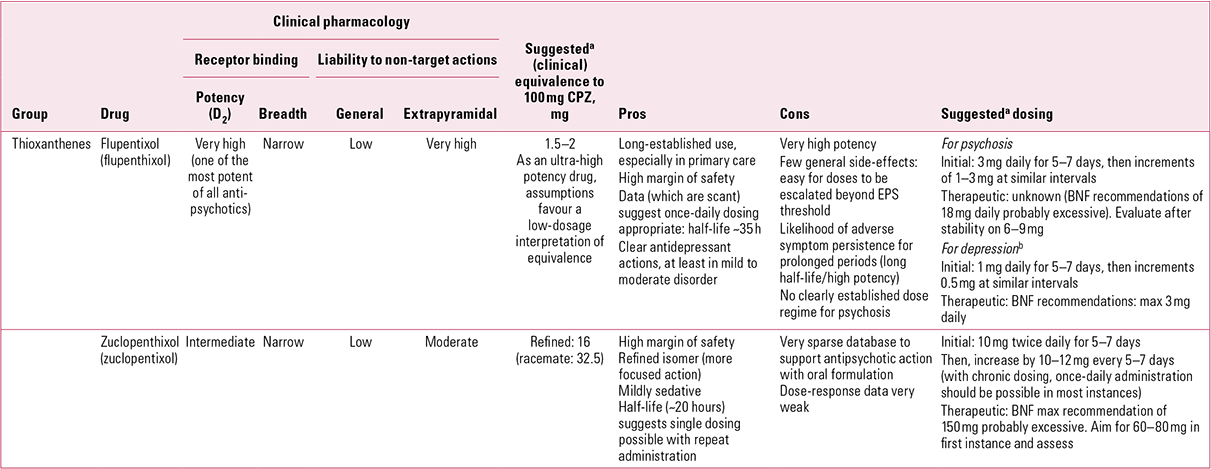
Flupentixol (along with fluphenazine) is among the most potent of all antipsychotics. This, combined with a relatively benign general adverse effect profile, makes it one of those antipsychotics most likely to be subject to unnecessary dose escalations. Minimum effective dose and true equivalence are therefore hard to assess. Usually assumed to be around 50 times more potent than chlorpromazine (Reference Atkins, Burgess and BottomleyAtkins 1997; Reference Gardner, Murphy and O'DonnellGardner 2010), in reality the figure may be closer to 100 times (Reference WyattWyatt 1976). Certainly, even taking a conservative view of its relative antipsychotic potency, the British National Formulary (BNF) recommendations of up to 18 mg/day for oral use still seem excessive (British Medical Association 2012). Its half-life (~19–39 h) makes it comfortably suitable for once-daily dosing (Reference JorgensenJorgensen 1980).
In addition to powerful D2 actions, flupentixol has anti-serotonergic (5-HT2) and anti-adrenergic (predominantly α1) actions, but its activity at cholinergic sites is too little to be clinically relevant (Fig. 1). It can therefore be associated with hypotension on first oral exposure and with extrapyramidal side-effects, though with gradual introduction and doses kept low, it is generally well tolerated.Footnote b
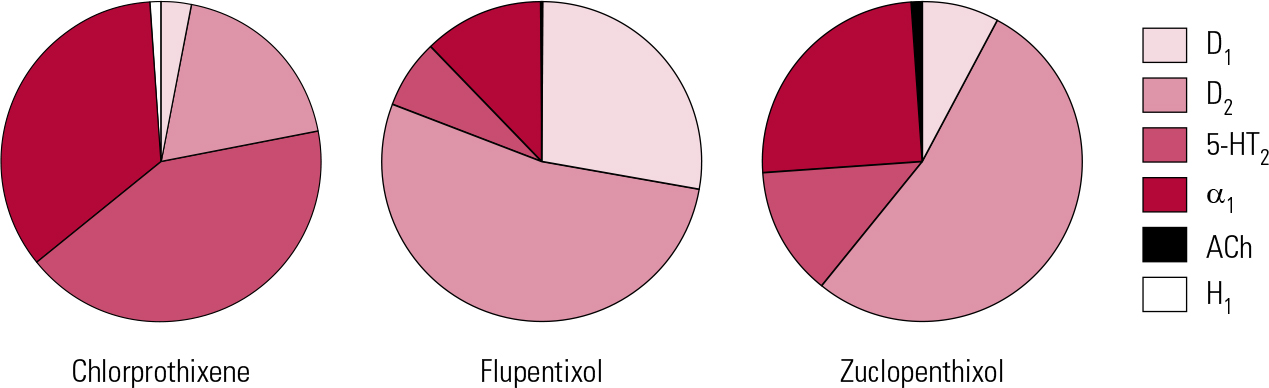
FIG 1 Receptor binding profiles of some thioxanthene antipsychotics by percentage of total binding contributed to by each transmitter type (after Reference Hyttel, Larsen, Christensen, Casey, Chase and ChristensenHyttel 1985).
Flupentixol has the greatest D1 antagonist activity of all the older antipsychotics. The significance of this is unclear for, clinically, it does not stand out as correspondingly different – apart from in one possible regard. Flupentixol has long been associated in low dose with antidepressant and anti-anxiety actions (Reference ReiterReiter 1969; Reference Hall and ColemanHall 1973; Reference Predescu, Ciurezu and TimoftePredescu 1973; Reference FrolundFrolund 1974; Reference Gruber and ColeGruber 1991). Most reports are anecdotal (e.g. Reference Trueman and ValentineTrueman 1974; Reference Becker, Grinberg and WeizmanBecker 2002) or find easy explanation in different dose regimes between flupentixol and comparator drugs (Reference Wistedt and RantaWistedt 1983), but a clinically impressive antidepressant action has been demonstrated in good trials. In a two-arm non-placebo-controlled study, Reference Young, Hughes and LaderYoung et al (1976) found that in mild to moderate depression, flupentixol had antidepressant efficacy comparable to that of amitriptyline, with greater anti-anxiety effects. In terms of quality of life, Reference Hertling, Philipp and DvorakHertling et al (2003) found that people with schizophrenia treated with flupentixol felt subjectively more relaxed and better able to deal with stress than those on risperidone.
We have seen similar claims for many early antipsychotics, especially when the search for marketable indications was active (Reference OwensOwens 2012, this issue). However, a reputation for beneficial mood actions must surely have stuck to flupentixol longest and most consistently – perhaps sufficiently so to accept the claim as valid. This is the only older antipsychotic with UK approval for use in depression.
In line with recent trends, such affective benefits have been attributed to serotonergic actions (Reference Becker, Grinberg and WeizmanBecker 2002), but autoreceptor effects – presynaptic D1 antagonism diminishing tonic inhibition of the transmitter synthetic enzyme tyrosine hydroxylase (or 3-monooxygenase) – highlighting as they do a prominent action of flupentixol, would seem worthy of consideration. However, the notion that flupentixol’s clinical pharmacology might allow it to be considered a ‘partial atypical’ antipsychotic (Reference Rachid, Bertschy and BondolfiRachid 2004) is to turn the taxonomical embarrassment of ‘atypicality’ (Reference OwensOwens 2008) seriously surreal.
Zuclopenthixol (zuclopentixol) (Table 1)
Clopentixol, the thioxanthene analogue of perphenazine, was introduced in the early 1960s, with the literature repeating an established pattern – therapeutic benefit in chronic schizophrenia unresponsive to other medications (Reference Ban, Ferguson and LehmannBan 1963; Reference Wolpert, Sheppard and MerlisWolpert 1967). As with flupentixol, it was originally presented as a racemic mix which, like flupentixol, has a striking impact on its pharmacology (Reference Johnstone, Crow and FrithJohnstone 1978). The pure trans (E) isomer, with minimal dopamine antagonistic activity, has little antipsychotic action, although it is highly sedative. Mixed racemates (e.g. clopentixol tablets, which have a trans:cis ratio of 2:1) share this sedative property, whereas purer cis isomers (e.g. Clopixol® depot, with a trans:cis ratio of 1:49) do so to a lesser degree (Reference NolenNolen 1983). Furthermore, while the pure cis isomer was found to have the same antipsychotic effect, on a mg/mg basis it was approximately twice as active as the trans (Reference Gravem, Engstrand and GulengGravem 1978). The purified cis isomer (subsequently renamed the Z isomer, incorporated in the marketed name, zuclopenthixol) was introduced to the UK in the late 1970s and is now the only form available.
Although refining the product increased potency and reduced the adverse effect profile, it did not abolish tolerability problems. Zuclopenthixol, perhaps because of its mild antihistaminic action, remains a more sedative preparation than flupentixol (even comparing depot formats) and, with little antimuscarinic activity, is prone to promote extrapyramidal side-effects (EPS) above a certain threshold, though these may be less persistent during acute treatment than those associated with haloperidol (Reference Heikkila, Eliander and VartiainenHeikkila 1992).
Unfortunately, that threshold is hard to determine as, once again, the dose-finding literature for oral formulations is poor, a particular problem when constituent components have changed. Older publications suggest chlorpromazine:clopentixol equivalence at between 4:1 and 5:1 (Reference WyattWyatt 1976; Reference Rey, Schulz and CostaRey 1989), though a more recent perception is of greater relative potency (~10:1; Reference Gardner, Murphy and O'DonnellGardner 2010). Some early trials utilised restraint in dosage (e.g. Reference Wolpert, Sheppard and MerlisWolpert 1967: 50 mg/day), but most studies on which equivalence tables are based used much higher doses (e.g. Reference Serafetinides, Collins and ClarkSerafetinides 1972: average 227 mg/day). The problem of older studies using racemic mixes in which sedative properties may have contributed to the therapeutic ‘package’ is insurmountable, and zuclopenthixol equivalence at, say, 8:1 (Reference Gardner, Murphy and O'DonnellGardner 2010) is largely guesswork. Certainly, the drug is best considered as of intermediate potency (doses in tens of milligrams per day; see Reference OwensOwens 2012, this issue), with efficacy likely at the lower end of BNF recommendations (i.e. ≤ 80 mg/day) and possibly as low as 60 mg (Reference Gardner, Murphy and O'DonnellGardner 2010). The BNF also places a single-dose restriction (40 mg) on zuclopenthixol (British Medical Association 2012). Although somewhat more active at noradrenergic sites than flupentixol, clinical experience would not suggest particular blood pressure concerns to justify such caution, especially with repeat dosing.
Unlike injectable formulations, oral zuclopenthixol has never enjoyed prescribing prominence with psychiatrists in the UK, for reasons that are not clear. Nonetheless, with a mildly sedative profile and wide dose range, tablet formats have probably been underutilised in specialist practice.
Butyrophenones (phenylbutylpiperidines)
Haloperidol, the foundation butyrophenone, was discovered through the pharmacologist Paul Janssen’s interest in the effects of physical properties on complex organic molecules. By subjecting pethidine (meperidine) to heat, he created nor- (or desmethyl-) pethidine, which when itself heated, transformed more or less to haloperidol. By 1988, the 30th anniversary of this elegant finding, haloperidol was the world market-leader antipsychotic in sales terms. What was not clear was how much its widespread use in US emergency rooms contributed to stellar sales, although by the late 1980s the shift in American psychiatry towards high-potency antipsychotics – and its detrimental consequences in terms of increased liability to extrapyramidal side-effects – had already been highlighted (Reference Baldessarini, Katz and CottonBaldessarini 1984). The prominence haloperidol still holds in US practice can be seen in more recent consensus equivalence recommendations (Reference Kane, Leucht and CarpenterKane 2003), which are based on this, and not chlorpromazine, as the standard.Footnote c There is, of course, no reason why haloperidol should not be considered the standard antipsychotic for reference purposes – provided one can be confident that this is a drug that clinicians have traditionally used thoughtfully, with restraint. The evidence does not support such an assumption (Reference OwensOwens 2008).
Haloperidol (Table 2)
Haloperidol was traditionally referred to as a ‘selective’ dopamine antagonist (Reference Hyttel, Larsen, Christensen, Casey, Chase and ChristensenHyttel 1985) and, although there is some truth to this, others can lay better claim to the title (see below). It is, however, active across the dopamine receptor family, and at D2 subtypes in particular produces among the greatest affinities of any antipsychotic. It has modest 5-HT2A actions and slightly greater affinity for α1 receptors (Fig. 2). As a result, it can be associated with hypotension, though this is usually mild, restricted to postural effects and rapidly habituates. For this reason haloperidol, along with other butyrophenones, has long been seen as suitable for parenteral administration, though the intravenous route should always be used with care and appropriate monitoring.
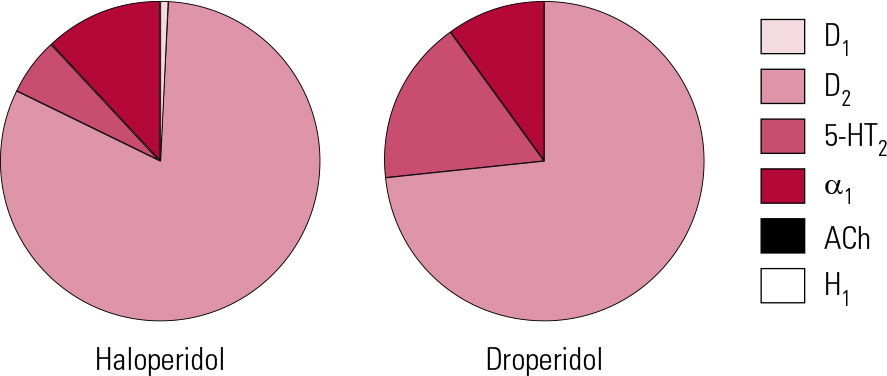
FIG 2 Receptor binding profiles of two butyrophenone antipsychotics by percentage of total binding contributed to by each transmitter type (after Reference Hyttel, Larsen, Christensen, Casey, Chase and ChristensenHyttel 1985).
TABLE 2 Non-phenothiazine older antipsychotics for oral use: butyrophenones
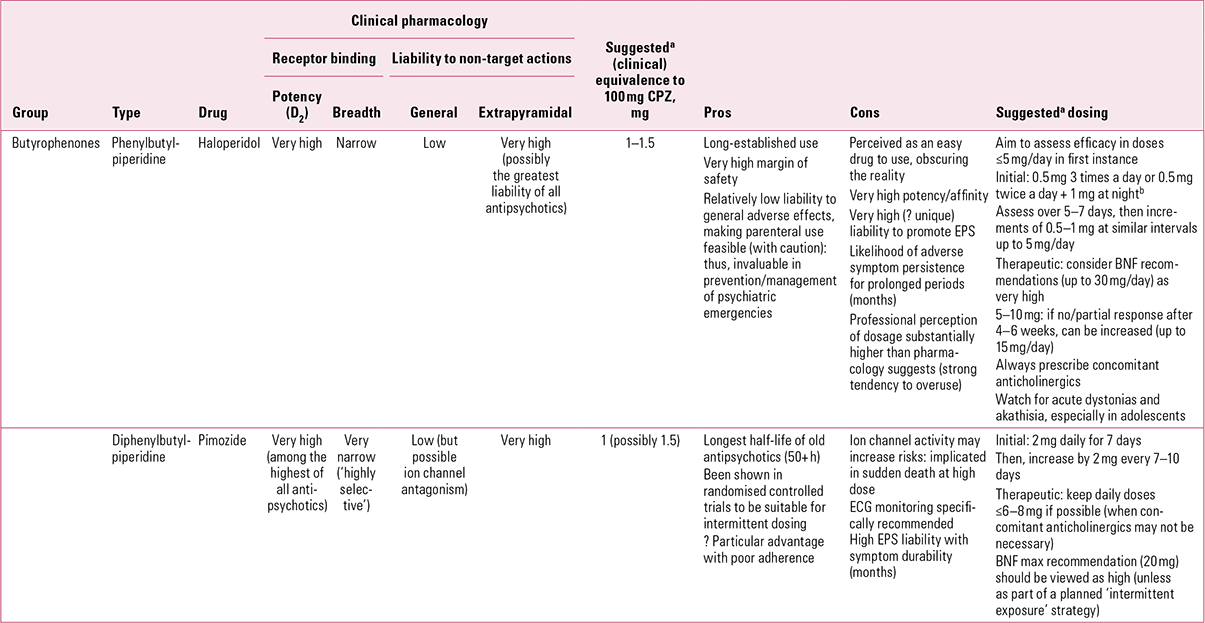
The drug has no significant antihistaminic or antimuscarinic actions (Reference Chew, Mulsant and PollockChew 2008). This lack of inherent anticholinergic activity could, in such a potent dopamine antagonist, account for a high liability to EPS and an accordingly poorer performance compared with other antipsychotics with broader binding profiles (Reference RosenheckRosenheck 2005). Haloperidol is, however, also among the most active compounds at sigma-1 (σ1) receptors (Reference Largent, Wikstrom and GundlachLargent 1987; Reference Cobos, Entrena and NietoCobos 2008) and, although the clinical significance of this remains unclear, it has been suggested that these receptors play a role in control of voluntary movement, especially through modulation of muscle tone.
Haloperidol is the high-potency antipsychotic par excellence, with all the advantages and disadvantages that come with that sobriquet. The advantages lie with a therapeutic index that is highly favourable – the drug retains a remarkable margin of safety over a seemingly limitless dose range. At the 30th anniversary meeting held in Antwerp in 1988, the pioneering psychopharmacologist Dr Frank Ayd caught his audience’s attention by describing a patient who received over 700 mg in 24 h! In translating a therapeutic index to practice, however, excellent safety becomes all too often confused with excellent tolerability – and therein lies the core of the disadvantages. Because of its limited receptor binding profile, haloperidol may be safe, but because of its exceptional anti-dopaminergic potency (and possibly actions at other sites), its EPS tolerability is very poor – perhaps uniquely so (Reference OwensOwens 1999) – making this, in my experience, a ‘difficult’ drug for doctors to prescribe.
This aspect of ‘difficulty of use’ can be related to evidence suggesting that, for maximal subjective experience, a very low dose in the range of 2.5 mg haloperidol achieves the ideal D2 occupancy (Reference de Haan, Bruggen and Lavalayede Haan 2003). These data support the view that subjective effects of antipsychotics are indeed extrapyramidally mediated and hence may reflect the inherent action of all compounds in the class (see Reference OwensOwens 2012, this issue).
Because haloperidol has traditionally been used in unnecessarily – sometimes cavalierly – high doses, standardising dose equivalences is difficult. Older American recommendations, based on studies at a time when restraint was more evident, suggested a chlorpromazine:haloperidol equivalence of about 40:1 (Reference WyattWyatt 1976). With the gradual increase in high-potency drug use, equivalence favoured a 20:1 ratio. This tended to be the benchmark adopted in most comparator studies for new antipsychotics. However, we know that therapeutic efficacy without EPS can be attained with doses lower than 5 mg/day (Reference Rosebush and MazurekRosebush 1999; Reference Oosthuizen, Emsley and TurnerOosthuizen 2001) and that such regimes are confirmed by functional brain imagining to achieve the desired D2 occupancy of < 80% %, thereby avoiding EPS (Reference Kapur, Zipursky and RoyKapur 1997). A relative potency of 50:1 can be inferred from Reference Davis and ChenDavis & Chen (2004), but even these authors acknowledge that 3.3–4 mg/day was the near-maximal effective dose range. Thus, it may be more appropriate to consider chlorpromazine:haloperidol relative potency as closer to 100:1, acceptance of which would demand a considerable shift in clinical practice.
Acute (single) dosing studies suggest that haloperidol has a half-life between 14 and 36 h (Reference Kudo and IshizakiKudo 1999), making it comfortably suitable for once-daily dosing. However, as with other antipsychotics, split dosing may be better for managing non-specific symptoms. With repeat dosing, however, the half-life of haloperidol can be extended considerably – in almost half of patients, to longer than 3 days (Reference de Leon, Diaz and Wedlundde Leon 2004). Cumulation may be another reason for the drug’s peculiar proneness to EPS. Like many, I previously advised that anticholinergics should be prescribed only if and when EPS emerged (Reference OwensOwens 1999), but I would now recommend, along with others (Reference RosenheckRosenheck 2005), that with high-potency, selective drugs such as haloperidol, anticholinergics should be automatically co-prescribed (Reference Owens, Johnstone, Owens and LawrieOwens 2010).
Butyrophenones are pharmacologically ‘much of a muchness’. Droperidol, with similar nor-adrenergic and somewhat greater 5-HT activity compared with haloperidol, was for many years a mainstay of emergency treatment in the UK. It fell for ‘commercial reasons’, but essentially on the back of the same QTc concerns that killed thioridazine (Reference Reilly, Ayis and FerrierReilly 2000, Reference Reilly, Ayis and Ferrier2002). The extent to which this reflected the drug per se or the emergency (and often restraint) circumstances in which it was used remains unclear (see below).
Benperidol, the only other butyrophenone available in the UK, has a similar profile to others of the group, although its antihistaminic and anti-adrenergic actions are evident only at high dose (Reference Schwarz, Hartung and LeuchtSchwarz 2005). Popular in some continental countries, especially Germany, it never found favour as an antipsychotic in the UK and is somewhat misclassified in the BNF. Here, its use has largely been restricted to controlling excessive or deviant sexual behaviour (as a so-called anti-libidinal) (British Medical Association 2012). It seems unlikely that its actions in this regard, poorly established anyway, are unique, while its cost (some 50 times that of the very similar haloperidol) is exorbitant for a drug on the market for over 40 years. Its persistence is probably an historical anomaly and its cost-effectiveness hard to envisage.
Diphenylbutylpiperidines
Only a relatively minor modification of the butyrophenone (or phenylbutylpiperidine) molecule produces the diphenylbutylpiperidines. These are therefore hardly a separate chemical group, although they are characterised by the longest half-lives of any of the older antipsychotics.
Pimozide (Table 2)
Pimozide can lay better claim to being a ‘selective’ D2 antagonist than haloperidol (Fig. 3). It is probably the most potent of the currently available antipsychotics, with powerful affinity for D2 receptors, in contrast to negligible D1 and muscarinic cholinergic actions. It does interact weakly at 5-HT2 and to a lesser extent α1 sites, though its affinity for histamine H1 receptors is minimal (Reference Sekine, Rikihisa and OgataSekine 1999). Furthermore, pimozide has significant calcium channel antagonist properties (Reference Galizzi, Fosset and RomeyGalizzi 1986), implicated in both favourable (mood elevating) and unfavourable (cardiac) actions. It is also one of only a handful of antipsychotics to inhibit biogenic amine transporters to a potentially clinically significant degree (Reference Tatsumi, Jansen and BlakelyTatsumi 1999).
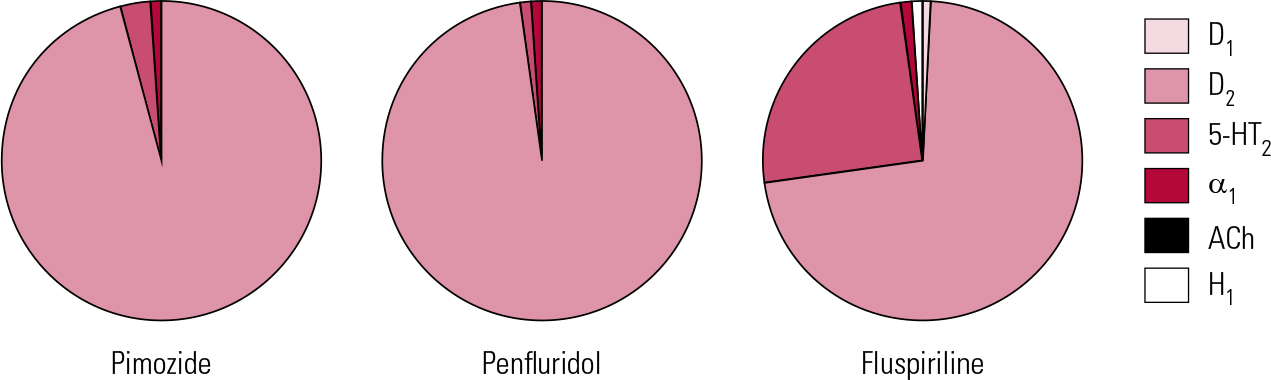
FIG 3 Receptor binding profiles of some diphenylbutylpiperidine antipsychotics by percentage of total binding contributed to by each transmitter type (after Reference Hyttel, Larsen, Christensen, Casey, Chase and ChristensenHyttel 1985).
Pimozide was one of a small number of drugs originally thought to be ‘novel’ (Reference OwensOwens 2008) and its use was initially targeted on ‘activating’ withdrawn individuals with chronic schizophrenia. Its blockade of transporter proteins (especially the serotonin and dopamine transporters), although less than that of antidepressants, might have accounted for an ‘alerting’ action, but yet again such action did not withstand closer scrutiny (Reference Sims and BurnsideSims 1975; Reference Wilson, Roberts and GerberWilson 1982). The proposal that pimozide could also be an effective treatment for tardive dyskinesia likewise fell, any benefits probably arising simply from suppression with the use of relatively large doses (Reference OwensOwens 1999) or masking by Parkinsonism (Reference Claveria, Teychenne and CalneClaveria 1975).
Even in the more restrained prescribing atmosphere of the 1970s, pimozide was considered to have a chlorpromazine-equivalent potency of approximately 80:1 (Reference WyattWyatt 1976), an assessment that has held (Reference Gardner, Murphy and O'DonnellGardner 2010), though this is not a drug licensed or much used as an antipsychotic in the USA. A more conservative assessment might rate its potency as high as 100:1 to 150:1. This, combined with a long half-life (~50 h, sometimes greatly extended to 150 h) is relevant to the fact that pimozide is the only orally formulated antipsychotic whose value has been demonstrated convincingly with other than daily use. Given on 4 days a week, Reference McCreadie, Dingwall and WilesMcCreadie et al (1980) found that it was as effective in maintenance as depot fluphenazine. In an intriguing study, it was then given once weekly to a group of in-patients using the same comparator (Reference McCreadie, Mackie and MorrisonMcCreadie 1982). On the basis of prior treatment regimes, doses ranged from 10 to 60 mg pimozide once weekly. Over a 9-month period, there was no difference in relapse rates between the groups. Blood monitoring showed a peak at 8 h, with return to more or less pre-administration levels after 4 days.
The study’s authors suggested that weekly treatment is genuinely ‘intermittent’ treatment. ‘Drug holidays’ (when treatment is temporarily suspended) were once popular for minimising exposure and reducing the risk of tardive dyskinesia but fell into disrepute when the opposite was demonstrated – namely, an increased risk of this side-effect (Reference Jeste, Potkin and SinhaJeste 1979). McCreadie and colleagues did record increased tardive dyskinesia ratings in their pimozide group, although this may have represented ‘withdrawal-emergent’ rather than the prognostically more sinister ‘treatment-emergent’ disorder. Two important findings were that elevations in prolactin were maintained only within a 48 h window post-administration, possibly suggesting lower hyperprolactin-related side-effects, and that substantial weight loss (on average 12 lb or 5.4 kg) occurred in the pimozide group.
Penfluridol and fluspirilene
This work highlights a long-held dream – an oral antipsychotic suitable for once-weekly maintenance. Penfluridol (Fig. 3) once encapsulated this but was withdrawn from most markets on safety concerns. As the above suggests, the value of such strategies might extend beyond patient preference to encompass real medical advantages. Pimozide was, however, one of the first antipsychotics to become ensnared in the ‘sudden death’ issue (Committee on Safety of Medicines 1990), with subsequent upper limits on daily dose (20 mg) recommended in the BNF. It is doubtful whether the doses in the above study (mean single weekly dose of 40 mg) would be acceptable now, at least without monitoring. The same principle may, however, be worth exploring with newer drugs with very long half-lives (e.g. aripiprazole).
A further unrealised dream is that of a specific antipsychotic for a specific mental state disorder, though pimozide still clings to its reputation of possessing particular efficacy in monosymptomatic hypochondriacal psychosis and related disorders (see the BNF). This view sprang largely from the work of a single Canadian group (Reference Riding and MunroRiding 1975a,Reference Riding and Munrob) and related to small numbers of patients. It lacks clinical credibility and, if one classifies disorders of this type as ‘delusional disorder’ variants, does not stand literature review either (Reference Manschreck and KhanManschreck 2006).
Fluspirilene (Fig. 3) was withdrawn in the UK a number of years ago but is of interest as being technically the first ‘long-acting injectable’ (LAI) (for the distinction between ‘LAIs’ and ‘depots’, see Reference Owens, Johnstone, Owens and LawrieOwens 2010). It is a very poorly investigated compound, but at approximately 3 weeks, its half-life appears to be the longest of any antipsychotic. It is water-insoluble and requires parenteral administration, but given weekly as an aqueous particle suspension is as effective as depots (Reference Abhijnhan, Adams and DavidAbhijnhan 2007). Its half-life suggests that an even longer interval between injections may be possible.
Substituted benzamides
Manipulations of the benzene ring have proved productive routes to drug development in many branches of medicine. Sulpiride, the most durable antipsychotic of the group, is a modification of the anti-emetic metoclopramide, itself a modification of procainamide.
Sulpiride (Table 3)
Sulpiride is the embodiment of the highly selective D2 antagonist approach to ‘anti-psychosis’ (Fig. 4). With no appreciable actions at D1 or D4 sites, limited actions at D3 (Reference StrangeStrange 2001) and only the weakest at 5-HT2A and α1 receptors (not clinically significant), it is the validation in practice of the ‘classical’ dopamine antagonist hypothesis of antipsychotic action. It may therefore seem perverse that it should be proposed by some as being ‘atypical’ (Reference Naber and LambertNaber 2009).
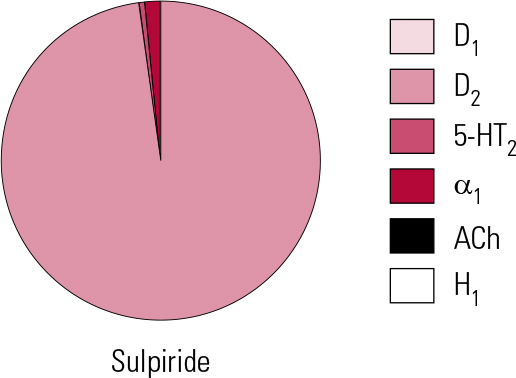
FIG 4 Receptor binding profile of the substituted benzamide antipsychotic sulpiride, by percentage of total binding contributed to by each transmitter type (after Reference Hyttel, Larsen, Christensen, Casey, Chase and ChristensenHyttel 1985).
TABLE 3 Non-phenothiazine older antipsychotics for oral use: substituted benzamides
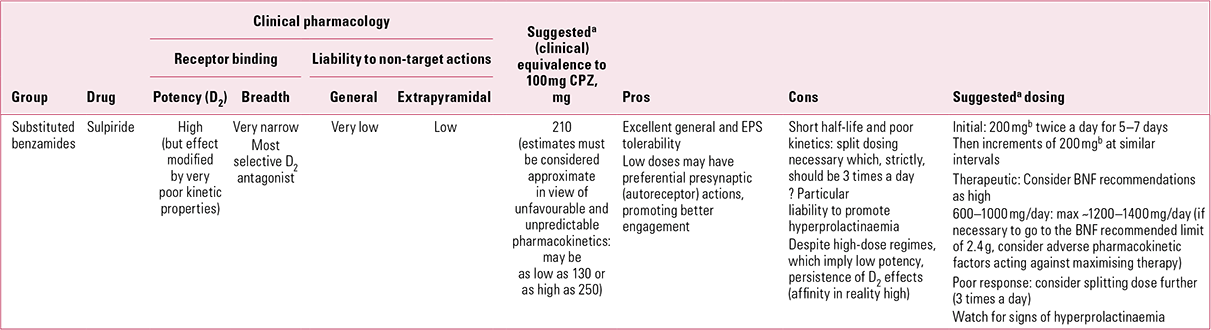
Elegant theories (e.g. limbic selectivity of action) have been constructed to explain this paradox, though an alternative pharmacokinetic explanation must not be discarded merely because it is simpler. In vitro D2 binding suggests that sulpiride should be considered of intermediate potency, yet clinically it must be used in high – for an antipsychotic, sometimes ultra-high – doses to achieve efficacy. Benzamide is a poorly soluble substance and, unlike most antipsychotics, sulpiride is predominantly water, not lipid, soluble. As a result, its low bio-availability (~30%) reflects poor absorption not, as with most antipsychotics, heavy presystemic metabolism and/or wide distribution (Reference Wiesel, Alfredsson and EhrneboWiesel 1980; Reference Owens, Stein and WilkinsonOwens 1998). Such absorption as does occur seems to take place via the clumsy mechanism of two sequential zero-order (i.e. saturable) processes (Reference Bressolle, Bres and Faure-JeantisBressolle 1992). That which is absorbed faces the further challenge of crossing the blood–brain barrier. It is to overcome these disadvantageous kinetic properties that high doses are required. This is a difficult drug to use in ‘dose equivalence’, as switches are likely to represent overall dose reductions from previous regimes, a point illustrated by the very low doses (~300–600 mg/day) now seen by specialists as appropriate (Reference Gardner, Murphy and O'DonnellGardner 2010). This is likely to be a significant reason for sulpiride still being considered ‘atypical’ – as it was 40 years ago (Reference OwensOwens 2008).Footnote d
Sulpiride’s unusual and poorly acknowledged kinetics is one factor underlying a lack of consensus on equivalent dosing. Chlorpromazine:sulpiride equivalence is usually taken as being in the range of 1:2 to 1:3 (Reference Rey, Schulz and CostaRey 1989; Reference Atkins, Burgess and BottomleyAtkins 1997), which would make an upper recommended dose of around 800–1200 mg/day (maximum ~1500 mg). However, this remains a drug about which clinicians show confusion. While estimating sulpiride to be only 75% as potent as chlorpromazine, experts then suggested an identical target dose range (300–600 mg/day) for the two (Reference Gardner, Murphy and O'DonnellGardner 2010). The very high upper dose recommended by the BNF (2.4 g/day; British Medical Association 2012) is likely to reflect a left-over from early studies, some of which utilised up to 3.2 g per day. The value of ‘mega-doses’ has not been demonstrated and must be suspect, especially in view of the saturable absorption mechanism.
Although calculated values are discrepant, sulpiride is a drug of relatively short half-life for an antipsychotic (~6–8 h) (Reference Jorgensen, Bridges and ChasseaudJorgensen 1986) and ideally should be given a minimum of three times daily. Despite this, twice-daily regimes have become the norm but on the basis of convenience/adherence rather than kinetics.
Sulpiride can be sedative in some patients, an action that does not find ready explanation in its known pharmacology nor, as it is largely excreted unchanged, in its metabolism. Moving beyond common receptor types, however, substituted benzamides interact with the γ-hydroxybutyrate (GHB) receptor (Reference Ratomponirina, Gobaille and HodeRatomponirina 1998), which may have a bearing on sedative actions. Sulpiride also produces higher prolactin levels than other, broader-spectrum antipsychotics (Reference Mannisto, Korttila and SeppalaMannisto 1978; Reference Harnryd, Bjerkenstedt and GullbergHarnryd 1984), something clinicians have long been aware of in increased rates of galactorrhoea. Extrapyramidal side-effects may not be the only non-target actions open to amelioration by broad receptor interactions, and brisk prolactin response may be the price that comes with high D2 selectivity. In this regard, the highest prevalence rates for hyperprolactinaemia have been reported with amisulpride (Reference Bushe, Yeomans and FloydBushe 2008), its close relative with even greater D2 affinity and dopaminergic selectivity.
Prescribing older antipsychotics: safety and tolerability
Antipsychotic prescribing, like all prescribing, involves an appraisal of risk run against benefit accrued (Reference OwensOwens 2008). At a population level, justification for antipsychotic use is easy. Untreated patients with schizophrenia have a reduced life expectancy compared with treated patients (Reference Tiihonen, Lonnqvist and WahlbeckTiihonen 2009). Seen through this lens, ‘benefit’ is so massive as to easily outweigh not only tolerability but even safety issues for the class as a whole. Clinicians, however, must practise through a more sensitive lens – that focused on the individual suffering an illness defined by its own individual characteristics. With the evidence now pointing to the irrefutable fact that antipsychotics do not differ meaningfully in either efficacy or effectiveness, tolerability becomes key to ‘tailored’ prescribing.
Some adverse (or ‘non-target’) effects have been mentioned, especially those relating predictably to receptor actions, such as sedation and hypotension. Overall, however, the list of potential tolerability issues is long (see Reference Owens, Johnstone, Owens and LawrieOwens 2010) and only a few significant concerns of greater relevance to older drugs will be raised here.
QTc and sudden death
Twenty-five years ago the American Psychiatric Association set up a working party to look at sudden death syndrome (SDS) occurring in individuals taking psychotropic medications (Reference Simpson, Jefferson and DavisSimpson 1987), though with antipsychotics the concern was longstanding (Reference Hollister and KosecHollister 1965). Although inconclusive, their report was followed by an expanding literature suggesting an association. A mechanism was subsequently outlined in prolongation of the QTc interval (the cardiac repolarisation phase corrected for heart rate), the risk emanating from the multifocal tachyarrhythmia, torsade de pointes. The pharmacological basis is believed to be blockade of the rapid component of the delayed rectifier potassium (IKr) channel.
It is now clear that patients receiving anti-psychotics can develop prolonged QTc (Reference Reilly, Ayis and FerrierReilly 2000; Reference Ray, Meredith and PurushottamRay 2001) and that all compounds share this potential. Furthermore, epidemiological studies support an increased risk of sudden death in those taking antipsychotics (Reference Reilly, Ayis and FerrierReilly 2002; Reference Ray, Chung and MurrayRay 2009). However, these facts do not necessarily tell prescribers all they need to know.
Although QT prolongation may be the best predictor of serious dysrhythmias, it remains a weak one. It is an even weaker predictor of sudden death (Reference ShahShah 2005; Reference SagerSager 2008), leaving the literature difficult to interpret. For example, in Reference Reilly, Ayis and FerrierReilly et al ’s (2002) sample, groups were not matched for exposure to diuretics, a major confound as hypokalaemia is known to prolong QTc (Reference Witchel, Hancox and NuttWitchel 2003); and although Reference Ray, Chung and MurrayRay et al (2009) established important dose–response relationships, the ranges devised bear little relationship to mainstream prescribing – unless antipsychotics are being used for non-specific purposes in patients vulnerable to QTc prolongation because of, for example, ischaemic heart disease.
Sudden death has been reported more often with older drugs, but one must again interpret this with caution as it largely reflects frequency of use rather than inherent risk. So-called atypicals are also implicated (Reference Ray, Chung and MurrayRay 2009). There is a further factor that can confound any apparent increased risk attributed to drugs, perhaps sufficiently powerful to explain the fact that with sophisticated analyses, independent effects of antipsychotics on QTc can be hard to demonstrate (Reference Ramos-Rios, Arrojo-Romero and Paz-SilvaRamos-Rios 2010). ‘Hyperarousal’, especially in situations where emergency tranquillisation or high-dose regimes are administered, can itself be associated with QT prolongation (Reference Bar, Koschke and BoettgerBar 2007). This is thought to result from decreased parasympathetic modulation, which in turn exposes the heart to unopposed sympathetic drive (‘sympathetic prolongation’). The loss of protective vagal modulation has been correlated with the severity of positive psychotic symptoms and duration of illness (Reference Bar, Letzsch and JochumBar 2005). The circumstances associated with administering antipsychotics may thus confound the relationship between them and sudden death, something more likely to ensnare older drugs, which remain the favoured compounds for controlling disturbed behaviour. Enforced restraint and tranquillisation should always be viewed as major undertakings with the potential for serious consequences.
An electrocardiogram (ECG) as part of routine medical assessment represents good psychiatric practice. However, if the focus is solely the QTc, there is no evidence that routine ECGs are effective in detection/prediction and they are certainly not cost-effective. Of far greater importance is a detailed history, including prescription of QT-prolonging drugs, and – crucially – family history of sudden premature death, heart disease and so on.
Cerebrovascular events in elderly people
In 2004, regulators received data from randomised controlled trials assessing the value of risperidone for the behavioural and psychological symptoms of dementia (BPSD) that suggested an unanticipated increase in cerebrovascular incidents. Concern extended to other new compounds, raising the question as to whether this was an issue with ‘atypicals’ or, of greater concern, an antipsychotic class effect.
Subsequent data have been contradictory and detailed exposition is beyond the scope of the present article. However, epidemiological findings do support an increased risk of cerebrovascular events, comparable with newer as with older drugs (Reference Trifiro, Spina and GambassiTrifiro 2009) and perhaps greatest in the early weeks of exposure (Reference Kleijer, van Marum and EgbertsKleijer 2009). The increased risk appears to hold when the focus is narrowed to stroke, though some findings suggest that the ‘atypicals’ do present a slightly enhanced risk (Reference Douglas and SmeethDouglas 2008). However, Sacchetti and colleagues found the risk to be greatest (some sixfold) with phenothiazines and general frailty as opposed to dementia (Reference Sacchetti, Trifiro and CaputiSacchetti 2008). These findings are worth noting as the study included data for stroke estimates in the unexposed elderly population that matched well with data from other international work, suggesting that the group captured a comprehensive sample. If true, this view weakens the perception of the likes of promazine as ‘benign’ and of especial utility for elderly people.
Careful consideration must be given to prescribing antipsychotics for older people, especially the frail, with decision pathways clearly recorded.
Venous thromboembolism
Venous thromboembolism in patients on anti-psychotics seems a lesser risk than was at one time thought, though recent work supports the view that low potency is a factor, especially in the 3 months after initiation (Reference Parker, Coupland and Hippisley-CoxParker 2010). The most likely explanation links thromboembolic events to weight gain, immobility, smoking and other lifestyle issues, but it remains possible that with phenothiazines (especially chlorpromazine), production of clot-promoting anticardiolipin antibodies is relevant.
Allergic and toxic reactions
Virtually all antipsychotics have been associated with isolated reports of allergic reactions, but the greatest risk lies with phenothiazines and, because of the ongoing frequency of its use, with chlorpromazine.
A typical hot, itching, erythematous skin rash coming on 10–14 days after first exposure and immediately on subsequent exposure affects up to 10% of patients. Hepatic dysfunction may show simply as deranged liver function tests or, in about 1% of individuals, frank jaundice. Although often described as allergically mediated, this cholestatic jaundice also comprises an element of toxicity (Reference Owens, Stein and WilkinsonOwens 1998). It is usually asymptomatic, although it may be associated with nausea, fatigue, itching, etc. and, very rarely, can progress to acute liver failure. A biliary cirrhosis, generally less sinister than the idiopathic variety, can also be a serious medical development.
Between 60 and 70% of patients on long-term chlorpromazine develop autoantibodies, especially antinuclear antibodies, and polyclonal immunoglobulin M (IgM) elevations are commonly seen with phenothiazines in general (Reference Canoso, De Oliviera and NixonCanoso 1990). The long-term significance of immunopathic changes is unclear and although chlorpromazine has been implicated in drug-induced systemic lupus erythematosus, this is very rare and usually relatively benign.
Phenothiazines in high dose over long periods can precipitate deposition of a melanin-like substance in soft tissues, including the skin, eyes and internal organs (oculocutaneous melanosis). Many antipsychotics can sensitise to sunlight, but again the major ‘culprit’ is chlorpromazine. This side-effect is a mixture of phototoxicity and photoallergy, with action in the UVA range, so brightness more than heat is what matters. It results from UV-induced dissociation of the chlorine ions which stimulate damaging free radical actions (Reference MooreMoore 2002). Barrier creams should be routinely available to those taking chlorpromazine and those on other phenothiazines who develop itching or burning.
Antipsychotics can also produce toxic suppression of bone marrow elements and, rarely, severe allergic failure. Compared with the detail with which clozapine’s effects on marrow have been charted, we know remarkably little about the long-term effects of older antipsychotics. However, even if routine monitoring reveals blood parameters within the normal or low-normal range, it is prudent to assume that in chronically treated patients, marrow is under stress with long-term antipsychotic exposure of any kind, a fact to bear in mind if clozapine-treated patients subject to polypharmacy suffer a fall in their neutrophil count and slip into ‘amber’ alert, when reducing/stopping all additional medication should be the priority.
Hyperprolactinaemia
Rises in serum prolactin have been attributed to all older antipsychotics, while much has been made of the newer drugs, where it seems ill-sustained or even absent. The likelihood of hyperprolactinaemia cannot, however, be used to categorise antipsychotics into ‘typical’ or ‘atypical’. Those newer drugs for which the phenomenon seems limited or absent are mostly of low potency with rapid dissociation characteristics (from D2) – essentially, clozapine and possibly quetiapine. Others are lower-potency compounds with broad binding profiles used in (relatively) low dose (e.g. olanzapine). A similar pattern would probably be expected with older drugs used with equal prudence. The clinical relevance of hyperprolactinaemia has been cogently argued (Reference Dursun, Wildgust and StricklandDursun 2008) but remains unclear.
Extrapyramidal side-effects
Extrapyramidal dysfunction slipped (or was pushed) down the list of perceived problems associated with antipsychotic drug use but – of course – never went away. Some ‘acute-end’ EPS, especially acute dystonias, have undoubtedly decreased in prominence with use of newer drugs. This is probably related to a mixture of pharmacological differences (e.g. lower potency/greater breadth of receptor interactions) and use in lower dose schedules than were previously in vogue for the older drugs. Differences in usage might also explain diminution in rates of tardive motor disorders, though reductions here remain to be established. However, the major conceptual and practical EPS issue for clinicians is Parkinsonism, because its pervasiveness and wide boundaries have never been clearly acknowledged and clinical means of delineating it from illness and other similar phenomena are without proven validity. Nonetheless, the evidence we have is that Parkinsonism is as prominent a risk as ever (Reference Miller, Caroff and DavisMiller 2008). The reader is referred to my previous article in Advances (Reference OwensOwens 2008) for why this remains of the utmost importance and to specialist sources for clinical accounts (e.g. Reference OwensOwens 1999).
The ‘minimum effective dose’
While everyone agrees that treatment with all antipsychotics should revolve around the ‘minimum effective dose’ (Tables 1–3), what that is and how to achieve it is usually unstated. With older antipsychotics, this is partly because we do not have the data necessary to craft dosage ‘algorithms’ in the face of highly variable kinetic and response characteristics, and partly because management policies act against ‘ideal’ treatment planning. In traditional environments, psychiatrists often operate under pressure to discharge early when the timeline for ‘improvement’ is in reality more protracted, a deficit they cover with dose escalation. In home or community contexts, however, pressure comes from ensuring that treatment is ‘adequate’, as much to cover perceptions of risk as to manage active symptomatology.
The intuitive but unsound principle underlying rapid dose escalations is that response times can be shortened accordingly. Systematic review has replicated early findings that greatest improvement in psychotic symptoms in schizophrenia occurs during the first week or two of treatment (National Institute of Mental Health Psychopharmacology Service Center Collaborative Study Group 1964; Reference Agid, Kapur and ArenovichAgid 2003). However, in my experience this reflects not fundamental improvement but non-specific benefits in anxiety and general arousal, removing some of the ‘drive’ fuelling psychotic symptomatology. There is experimental support for such a view (Reference Johnstone and SimisterJohnstone 1979). This being the case, the first priority is to avoid rapid dose escalations during the first week or two of exposure, especially since this is when kinetic parameters are changing to those of repeat exposure, which themselves may facilitate tolerability. It is also important to bear in mind that, pharmacologically, the largest dose increment of all is the one from zero to any starting regime, no matter how modest.
Thus, the key to maximising tolerability and ensuring minimum exposure with antipsychotics lies in the first few days of treatment.
One notion that has, for a second time, fallen into disregard is the ‘neuroleptic threshold’. This was based on the simple, intuitive idea that once a certain D2 occupancy is passed one has moved beyond the necessary therapeutic dose into the realms of side-effects and that this transition can be detected clinically by the presence of subtle extrapyramidal signs, the most widely discussed being changes in handwriting (Reference HaaseHaase 1961). Once these, or any clinical markers of extrapyramidal dysfunction, emerge cautious reduction of dose should maximise the likelihood of benefit while minimising EPS risk.
Imaging work supports this principle (Reference Farde, Norstrom and WieselFarde 1992), and in the early 1990s these ideas were revived only to sink again with the introduction of new agents, with their unjustifiable reputation for beneficence. The variability of handwriting makes reliability an issue, but the principles of not increasing to a new dose until the likelihood of EPS emerging can be assessed on the existing dose, and not pushing doses beyond those that precipitate EPS, is a good framework for sound practice. Prescribers are encouraged to view BNF recommendations for escalations (e.g. every 3 days or so) as, in most instances, representing the ‘rapid’ end of the spectrum (British Medical Association 2102).
Conclusions
The use of older antipsychotics is not inherently more ‘difficult’ or more ‘risky’ than newer drugs. Perhaps the ‘difficulty’ with antipsychotics lies in trying to dispel the myths of the past two decades – that there is a group of these compounds inherently ‘better’ than what came before. The only thing inherent to antipsychotic drugs is that, as a class, they are all difficult to use – both for doctors who prescribe them and for patients who take them. Seeing antipsychotics as challenging therapeutic agents is the first step in acquiring the skills that utilising them to maximum effect demand and that should be part of every psychiatrist’s core competencies.
MCQs
Select the single best option for each question stem
-
1 Of the following, the longest half-life is held by:
-
a flupentixol
-
b fluspirilene
-
c haloperidol
-
d pimozide
-
e zuclopenthixol.
-
-
2 ‘Highly selective’ dopamine D 2 antagonist applies most accurately to:
-
a flupentixol
-
b haloperidol
-
c pimozide
-
d sulpiride
-
e zuclopenthixol.
-
-
3 QTc prolongation is a consequence of blockade of:
-
a Cav2 calcium channels
-
b GABA–chloride ionophore channels
-
c KCa calcium-activated potassium channels
-
d IKr potassium channels
-
e Nav1 sodium channels.
-
-
4 On the basis of a once-weekly dosing schedule, pimozide has been found to be associated with:
-
a comparable maintenance effect to long-acting injectable risperidone
-
b no tardive dyskinesia
-
c post-absorptive blood level peak (Cmax) delayed until day 4
-
d prolactin levels sustained over the 7-day cycle
-
e significant weight loss.
-
EMI
This question relates to both parts this article
Theme: Antipsychotic therapeutics
Options
-
a Chlorpromazine
-
b Haloperidol
-
c Flupentixol
-
d Levomepromazine
-
e Pericyazine
-
f Perphenazine
-
g Pimozide
-
h Prochlorperazine
-
i Promazine
-
j Sulpiride
-
k Trifluoperazine
-
l Zuclopenthixol
Link up the most appropriate drug option(s) above with the statements below
-
1 in psychotic states, is particularly useful for sedative properties (choose 3)
-
2 is most likely to be associated with hyperprolactinaemia (choose 1)
-
3 is a moderate inhibitor of CYP2D6 (choose 2)
-
4 is approved in the UK for short-term/adjunctive use in severe anxiety (choose 5)
-
5 is approved in the UK for use in depressive states (choose 1).
MCQ answers
| 1 | b | 2 | d | 3 | d | 4 | e |
EMI matchings

| 1 | a,d,e | 2 | j | 3 | b, f | 4 | a, e, f, h, k | 5 | c |








eLetters
No eLetters have been published for this article.