

It has been argued that schizophrenia is the most prevalent neuropsychiatric illness, understood as a debilitating neurodevelopmental disorder that appears at a critical period in early adulthood (Reference Frangou and MurrayFrangou 2000). In recent years, there has been increasing interest in the study of prodromal stages of this disorder and its evolution into clinically significant psychosis, the acceptance of a ‘stress–vulnerability model’ and the combined role of genetic and environmental influences (Reference Oh and PetronisOh 2008). However, the underlying pathophysiological mechanism by which the environment influences risk of psychosis remains essentially unproven. The vast majority of biological research into the pathogenesis of schizophrenia has focused on neurotransmitter abnormalities, with central structural changes also apparent. Yet several findings, both historical and more recent, suggest the involvement of immunological processes as well (Reference Aghajanian and MarekAghajanian 2000; Reference Howes and KapurHowes 2009).
A brief history
Dameshek published a first study of white blood cells in dementia praecox in 1930 (Reference DameshekDameshek 1930) and some 30 years later Saunders & Muchmore wrote on the effects of phenothiazine antipsychotics on the immune system (Reference Saunders and MuchmoreSaunders 1964). Further evidence implicating the immune system and suggesting a viral aetiology of schizophrenia was reported in 1985, with elevated risk of schizophrenia in people born in the winter months (Reference Bradbury and MillerBradbury 1985), a finding confirmed in wide epidemiological studies at the end of the 20th century (Reference Mortensen, Pedersen and WestergaardMortensen 1999). In the 1990s, the first suggestions of an autoimmune aetiology were proposed (Reference Wright, Takei and RifkinWright 1995), again mediated by a prenatal viral exposure.
More recently, our understanding of the complexity of the immune system at a cellular level, including the identification of phenotypically and functionally distinct T helper cell subsets, the influence of medication on immune function and the genetics underlying immune-related illness, has led to a resurgence of interest in this area, with several key findings. Although dopaminergic dysfunction in schizophrenia is clearly demonstrated, the underlying mechanisms for this may well lie in the glutamate system, itself affected by immune function (Reference Schwarz, Gerlach, Deckert and DoubleSchwarz 2007). Difficulties in previous studies of immune dysfunction in schizophrenia, in terms of replication of results and heterogeneity of populations, have begun to be overcome by large-scale meta-analysis, with results indicating that this mostly neglected area may be highly significant (Reference MüllerMüller 2011).
Box 1 explains some key terms used in the article.
BOX 1 Some key terms
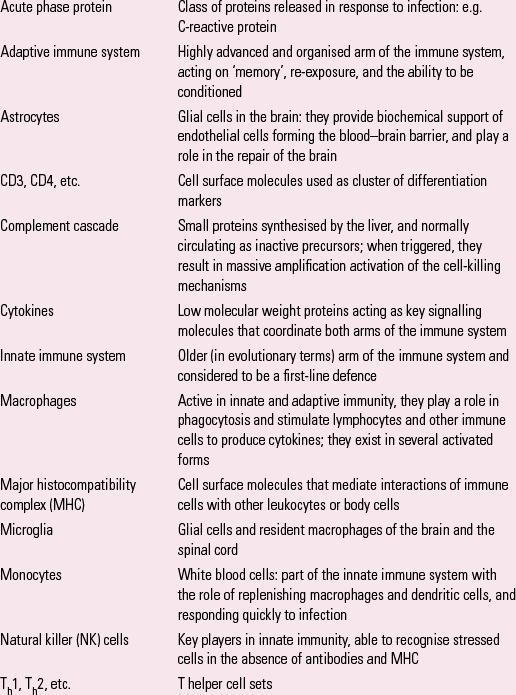
| Acute phase protein | Class of proteins released in response to infection: e.g. C-reactive protein |
| Adaptive immune system | Highly advanced and organised arm of the immune system, acting on ‘memory’, re-exposure, and the ability to be conditioned |
| Astrocytes | Glial cells in the brain: they provide biochemical support of endothelial cells forming the blood–brain barrier, and play a role in the repair of the brain |
| CD3, CD4, etc. | Cell surface molecules used as cluster of differentiation markers |
| Complement cascade | Small proteins synthesised by the liver, and normally circulating as inactive precursors; when triggered, they result in massive amplification activation of the cell-killing mechanisms |
| Cytokines | Low molecular weight proteins acting as key signalling molecules that coordinate both arms of the immune system |
| Innate immune system | Older (in evolutionary terms) arm of the immune system and considered to be a first-line defence |
| Macrophages | Active in innate and adaptive immunity, they play a role in phagocytosis and stimulate lymphocytes and other immune cells to produce cytokines; they exist in several activated forms |
| Major histocompatibility complex (MHC) | Cell surface molecules that mediate interactions of immune cells with other leukocytes or body cells |
| Microglia | Glial cells and resident macrophages of the brain and the spinal cord |
| Monocytes | White blood cells: part of the innate immune system with the role of replenishing macrophages and dendritic cells, and responding quickly to infection |
| Natural killer (NK) cells | Key players in innate immunity, able to recognise stressed cells in the absence of antibodies and MHC |
| Th1, Th2, etc. | T helper cell sets |
An overview of the immune system
The body’s defence against threatening microorganisms, the immune system, is much more complex and differentiated than first imagined. The immune response is a highly coordinated process involving an array of cell types, normally protecting the body from pathogens while maintaining tolerance of self-antigens and harmless environmental antigens. Conceptually, the immune system is traditionally divided into two arms – the innate immune system and the adaptive immune system – each with subsequent cellular and humoral components. There is considerable ‘cross-talk’ between the two arms.
The innate immune system
The first arm comprises our innate defence mechanisms, older in evolutionary terms and considered to be a first-line defence. Its cellular components consist of monocytes and macrophages, dendritic cells and natural killer cells. These recognise pathogens and promote defence against them, but lack the sophistication to adapt that is a benefit of more recent additions to the immune system. The innate humoral or antibody-mediated component comprises: acute phase proteins (e.g. C-reactive protein); the complement cascade, which allows phagocytic cells to clear pathogens from an organism; and various cytokines.
The adaptive immune system
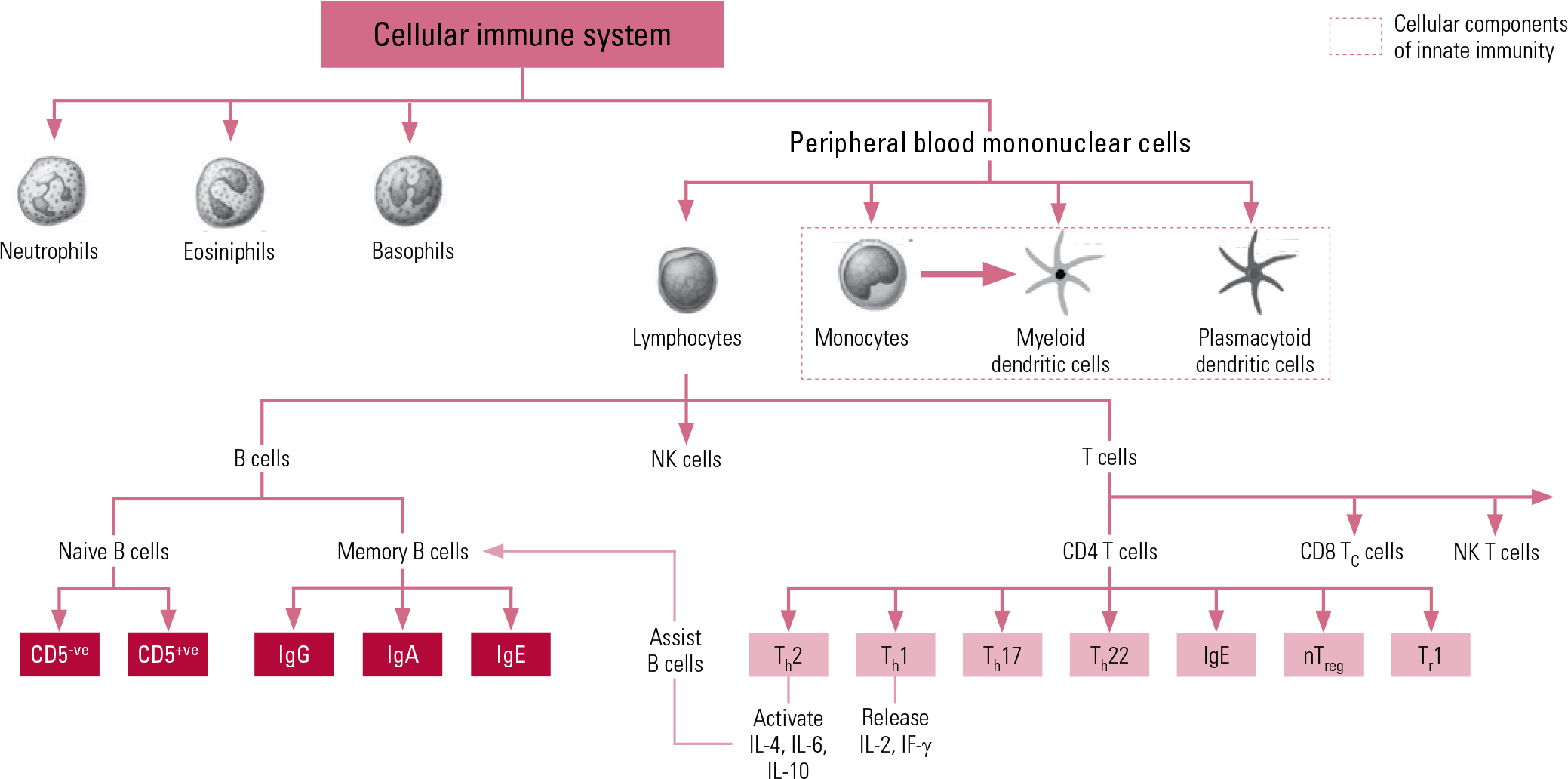
The second arm of our immune system is the adaptive system, which is more highly advanced and organised, acting on ‘memory’, re-exposure and the ability to be conditioned. The prime cellular components of the adaptive system are T lymphocytes and B lymphocytes, known more simply as T cells and B cells. The T cells comprise key components of the T helper 1 (Th1) system and the T helper 2 (Th2) system. The Th1 system produces pro-inflammatory (activating) cytokines such as interleukin-2 (IL-2), interferon-gamma (IF-γ) and tumour necrosis factor-alpha (TNF-α). The Th2 system promotes the generation and maintenance of antibody-mediated immune responses as well as production of anti-inflammatory cytokines such as IL-4, IL-10 and IL-13. The humoral component of the adaptive system includes these various circulating cytokines and specific antibodies. More recently recognised additions to the adaptive immune system include regulatory T cells (‘suppressor’ T cells) and other specialised T helper cells (e.g. Th17 and Th122). Figure 1 shows a schematic representation of cellular immunity.
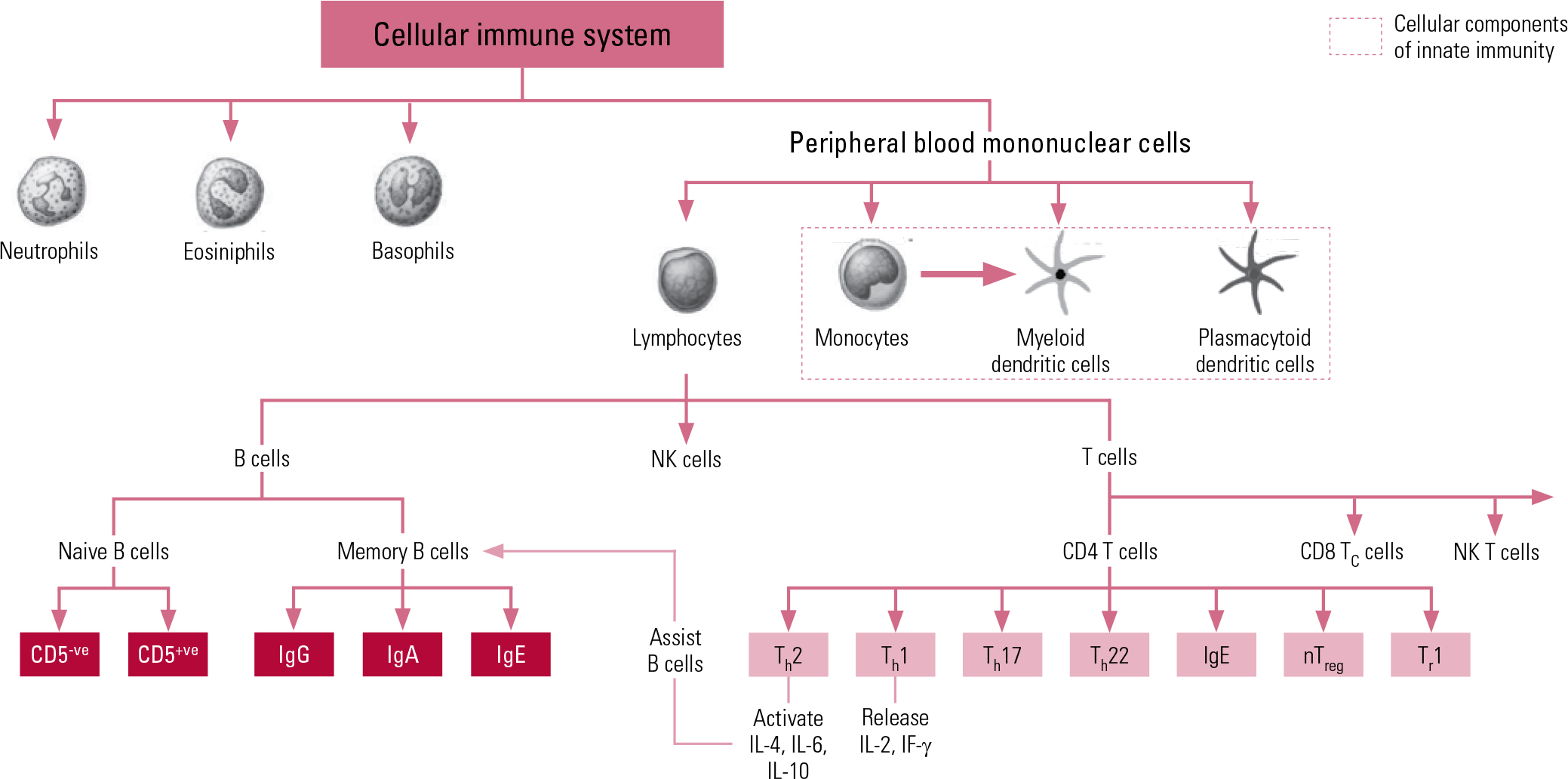
Fig 1 Schematic representation of components and interactions of cellular immunity. CD, cluster of differentiation marker; Ig, immunoglobulin; IF, interferon; IL, interleukin; NK, natural killer; nTreg, natural regulatory T cell; Th, T helper cell; Tr, regulatory T cell.
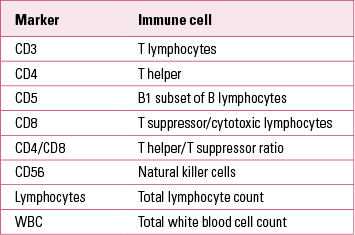
Markers of immune active cells and their parameters are used to measure total levels of different immune cells in peripheral blood, and are useful in clinical practice in immune and autoimmune disorders. Cell surface molecules (Table 1) are used as cluster of differentiation (CD) markers in the phenotyping of cells in the differential blood count (Reference Müller and SchwarzMüller 2010a).
TABLE 1 Common cell surface markers
Cytokines and their function
Cytokines and other immune molecules are essential in the development and function of the central nervous system, and are core actors in the maintenance of neuronal integrity, neurogenesis, synaptic remodelling and neurotransmission (Reference Raison and MillerRaison 2013). Often cited in literature investigating schizophrenia, cytokines are low molecular weight proteins that act as key signalling molecules to coordinate both arms of the immune system and exert direct effects in the periphery of the body and the brain. They can cross the blood–brain barrier and activate astrocytes (Reference Müller, Myint and KrauseMüller 2013). Cytokines can stimulate or inhibit the differentiation, proliferation or function of immune cells and are also thought to be important mediators of the ‘cross-talk’ between the central nervous system and immune system (Reference Potvin, Stip and SepehryPotvin 2008).
Immune hypotheses of schizophrenia
The cytokine theory of schizophrenia proposes that, in individuals who are genetically predisposed to schizophrenia, exposure to maternal viral infection during fetal life subsequently interferes with normal brain development, mediated through an immune response to this infection (Reference Sham, O'Callaghan and TakeiSham 1992). Animal studies indicate that it may not be exposure to a particular virus but the subsequent immune response that determines risk (Reference MüllerMüller 2011).
As mentioned above, cytokines are essential for synaptogenesis during early development and in periods of neuronal plasticity in adult life. Disruption of the relative balance of cytokines or microglial activation, for example by early maternal infection, may have direct effects but also more nuanced changes visible later in development (Reference Monji, Kato and KanbaMonji 2009). Infectious agents such as the influenza virus, borna virus, chlamydia and toxoplasma gondii have been suggested as risk factors for schizophrenia (Reference Müller and SchwarzMüller 2010a). These diverse viral agents and infections may have their influence directly in the developing fetal brain through a common mechanism of the maternal immune response. Prenatal exposure to an early inflammatory response may also cause a priming of immune reactions, leading to a more vigorous or atypical response to later environmental challenges. Thus, the prenatal exposure continues to exert effects well past the early developmental period and into late adolescence, a time of heightened neuronal plasticity and change, bringing the development of illness (Reference Fineberg and EllmanFineberg 2013).
The common and increasingly recognised occurrence of affective symptoms in schizophrenia (Reference UpthegroveUpthegrove 2009) may also be explained by shared immune and inflammatory processes, including an increase in cytokines and immune response (Reference Fineberg and EllmanFineberg 2013).
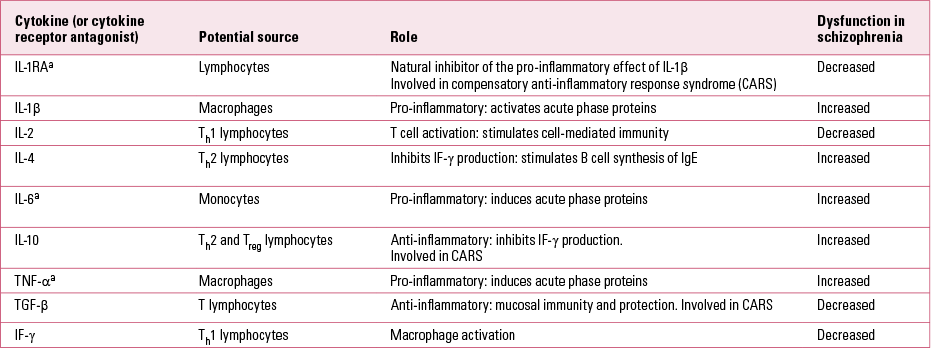
The current evidence for disruption as part of this abnormal immune response (Table 2) can be further grouped into three main hypotheses: chronically activated macrophages and T lymphocytes; imbalance in the T helper system; and the microglial hypothesis.
TABLE 2 Cytokines implicated in the pathogenesis of schizophrenia
Chronically activated macrophages and T lymphocytes
It is proposed that chronically activated macrophages, microglia and T lymphocytes (T cells) release inflammatory cytokines that over time result in the neuronal changes we see in schizophrenia. Monocytes carrying CD54 markers (intracellular adhesion molecules) bind to intracellular molecules, leading to the migration of further monocytes through endothelial cells in the blood vessels (Reference Cazzullo, Sacchetti and GalluzzoCazzullo 2001). Recently, Reference Miller, Gassama and SebastianMiller et al (2013) showed in a systematic review that in patients with active psychosis the total white blood cell count and proportion of CD4 (T helper), CD8 (T suppressor) and CD56 (natural killer) lymphocytes were significantly increased. A similar effect, with an increase in CD4/CD8 ratios, was seen in drug-naive first-episode psychosis, suggesting this is not a medication effect. In stable medicated patients, a significantly lower CD3 level, as a percentage of the total white blood cell count, was shown. It has been suggested that chronic excessive cytokine secretion could be the cause of the neurotransmitter dopamine and gamma-aminobutyric acid (GABA) abnormalities seen in schizophrenia (Reference Smith and MaesSmith 1995). Treatment with antipsychotic medication may normalise the CD4/CD8 ratio. Thus, there is a proposal that schizophrenia can be explored through a state marker of CD4/CD8 balance and a trait marker of CD56 (Reference Miller, Gassama and SebastianMiller 2013).
Imbalance in the T helper system
Patients with psychosis may have an imbalance between the normal Th1 and Th2 arms (Reference Müller, Krause and DehningMüller 2010b), with a dominance of the Th2 antibody-dependent response. A consistent finding is a decrease of IL-2 and IF-γ, demonstrating a diminished Th1 response. Also found is a reduced response after stimulation with antigens (Reference Müller and SchwarzMüller 2010a) and evidence for an increase in TNF-α (Reference Pedrini, Massuda and FriesPedrini 2012). In contrast, an activation of the Th2 immune response has been seen, including an increase in immunoglobulin E (IgE; an immunoglobulin often associated with response to allergies), IL-4 and IL-10. Studies looking at cerebrospinal fluid indicate that_the overactivated Th2 response is not just a peripheral phenomenon. A meta-analysis of data from 62 studies with a total sample of 2298 people with schizophrenia and analysis of 10 cytokines showed a significant increase in IL-1 receptor antagonists (IL-1RA), IL-2 receptors (IL-2R) and IL-6 and a decrease in IL-2 in schizophrenia. No significant effect sizes were obtained for the other cytokines, contradicting the hypothesis of Th2 dominance. IL-1RA and IL-6 are products of the innate immune system, and Reference Potvin, Stip and SepehryPotvin et al (2008) suggest that a primary alteration in this arm of the immune system may be at fault.
The microglial hypothesis
Inflammatory cytokine activators such as IF-γ activate microglia in the central nervous system. Microglia are the mononuclear phagocytes of the brain and play a major role in brain development, synaptogenesis and synaptic pruning. Activated microglia release further pro-inflammatory cytokines. These mediators can cause neuronal degeneration, white matter abnormalities and decreased neurogenesis. Reference Monji, Kato and KanbaMonji et al (2009) have proposed that a hypoglutamatergic state and impaired N-methyl-D-aspartate (NMDA) signalling are present in schizophrenia as a result of this action. NMDA receptor antagonists such as ketamine induce microglial activation and are well known to produce positive symptoms of psychosis. Atypical antipsychotics have been shown to decrease serum levels of cytokines such as IL-6 and TNF-α, and therefore it is suggested that some of their effectiveness may be mediated through an anti-inflammatory action on microglial activation (Reference Miyaoka, Yasukawa and YasudaMiyaoka 2007).
Challenges to immune hypotheses
Confounding factors in research investigating the immune system are multiple and often uncontrolled for in older studies. Medication is the main confounder, and it has been repeatedly shown to have a significant effect on the immune system (Reference Gordon and BarnesGordon 2003). Indeed, suggestions are that antipsychotic medication may in part be acting through a ‘balancing effect’ on cytokine functions. Several studies demonstrate the correlation between starting antipsychotic mediation and a decrease in IL-6. Some studies have also shown an increase in TNF receptors and plasma CD8, IL-6 and IL-1RA concentrations during clozapine therapy, although it is possible that this effect may in part be related to weight gain (Reference Maes, Bosmans and KenisMaes 1997; Reference Reichenberg, Yirmiya and SchuldReichenberg 2001; Reference Kluge, Schuld and SchachtKluge 2009).
Some studies have looked at drug-free or medication-naive patients, but the majority of research has investigated patients with established schizophrenia and a ‘drug-free’ or ‘recently commenced on medication’, rather than a truly antipsychotic-naive, status. To reduce the role of this major confounding factor, it may be important to study changes in cytokine levels during the first psychotic episode in drug-naive patients. In one of the few studies to include drug-naive participants, Reference Xiu, Chen and WangXiu et al (2012) found that levels of serum IL-18, a pro-inflammatory cytokine, were significantly increased in patients on antipsychotic medication for chronic schizophrenia compared with drug-naive patients with first-episode psychosis and healthy controls, and also that IL-18 levels were positively correlated with severity of symptoms.
The autoimmune hypothesis of schizophrenia
Schizophrenia occurs more frequently in families with a history of autoimmune disorders, for example psoriasis, Graves’ disease and coeliac disease. The exception is rheumatoid arthritis, where there is an inverse association. The presence of auto immune disease increases the risk of schizophrenia by 29% (Reference Benros, Nielsen and NordentoftBenros 2011) and systemic lupus erythematosus is associated with neuropsychiatric symptoms in the majority of patients (Reference Wekking, Nossent and van DamWekking 2010).
The Th1/Th2 imbalance hypothesis suggests that in schizophrenia there is overactivity of the autoantibody-producing arm of the immune system. Taken together with recent discoveries in autoimmune encephalitis, there is a compelling case for further study here.
What is autoimmune encephalitis?
‘Autoimmune encephalitis’ has been used interchangeably with the term ‘limbic encephalitis’, coined by Corsellis in 1968. It describes a neuropsychiatric syndrome characterised by subacute onset of memory disturbance, hallucinations and seizures, with evidence of inflammation in the medial temporal lobes (Reference Corsellis, Goldberg and NortonCorsellis 1968).
Traditionally, autoimmune encephalitis was thought to present only with a combined occurrence of psychiatric and core neurological symptoms: a first stage of confusion, amnesia and seizures, followed by fluctuating consciousness, autonomic dysfunction and dyskinesia. Initially, an underlying neoplasm was thought to account for the vast majority of cases, and psychiatric symptoms were thought to be brief and not dominant in the clinical picture as a whole (Reference Rickards, Jacob and LennoxRickards 2014, this issue). In the past 5 years, however, there has been an increase in the ability to diagnose autoimmune encephalitis, with a number of assays detecting autoantibodies responsible for encephalitis identified. These include synaptic proteins such as NMDA receptor and GABA receptor (GABA-BR1 and GABA-BR2) antigens, and cell surface antigens such as voltage-gated potassium channel (VGKC) antigens (Reference Irani and VincentIrani 2011; Reference Zandi, Irani and LangZandi 2011). NMDA receptor encephalitis in particular is now recognised to occur in the absence of other overt neurological symptoms. In case report studies, neoplasms have been shown to be present in only 20–50% of patients (Reference Zandi, Irani and LangZandi 2011). Additionally, in the past 18 months it has been proposed that a proportion of all psychotic illness, including that in patients presenting to mental health services with first-episode psychosis, may be antibody mediated (Reference Lennox, Coles and VincentLennox 2012; Reference Tsutsui, Kanbayashi and TanakaTsutsui 2012).
Treatments for autoimmune encephalitis include immunomodulation, intravenous immunoglobulin, plasma exchange and other immunosuppressive therapies. Response is seen within weeks, with relief of all symptoms achievable in the vast majority of patients (Reference Titulaer, McCracken and GabilondoTitulaer 2013).
NMDA receptors as pathogenic pathway in schizophrenia
The neuropsychiatric presentation of autoimmune encephalitis also provides evidence for the possibility of NMDA receptor function as a pathogenic pathway in schizophrenia itself. The NMDA receptors are excitatory cation channels with an essential role in synaptic transmission and neuronal plasticity, also with distinct receptor subtypes with pharmacological properties. We know that overactivity of NMDA receptors may provide a mechanism for epilepsy; conversely, low activity induces symptoms similar to schizophrenia (Reference CoyleCoyle 2006; Reference Dalmau, Gleichman and HughesDalmau 2008). Thus, the relation between anti-NMDA receptor encephalitis and schizophrenia has been described as ‘two sets that intersect, resulting in a common subset’ (Reference van de Riet and Schieveldvan de Riet 2013). Some authors have recently suggested that up to 10% of cases of first-episode psychosis have an autoimmune aetiology (Reference Lennox, Coles and VincentLennox 2012; Reference Tsutsui, Kanbayashi and TanakaTsutsui 2012). However, it is also clear that the vast majority of patients with confirmed autoimmune encephalitis have psychiatric symptoms, predominantly psychosis, as the main presenting symptom. Reference Dalmau, Gleichman and HughesDalmau et al (2008) reported that over 80% of patients with autoimmune encephalitis first presented to psychiatric services.
Stress and immunity
Our immune response to stress is considered to be largely mediated through the actions of cortisol. This steroid hormone belonging to the glucocorticoid groups is produced by the adrenal gland and secreted during a stress response. The primary function of cortisol is to redistribute glucose to regions of the body required for a fight-or-flight response, but it also acts to suppress the immune system. During chronic stress, cortisol is overproduced, causing alteration in immune functioning. We know that stress plays an important role in the onset and course of psychosis and that life events often precipitate first onset and relapse (Reference Frangou and MurrayFrangou 2000). High expressed emotion in patients’ families, resulting in stress and relapse of illness, is one of the most consistent findings in the course of schizophrenia (Reference Upthegrove, Atulomah and BrunetUpthegrove 2012). Patients with schizophrenia, contrary to previous beliefs, are particularly vulnerable to the stresses of everyday events (Reference Myin, Peeters and HavermansMyin 2003).
The autonomic nervous system (ANS) and hypothalamic–pituitary–adrenal (HPA) axis are important biological systems involved in stress processing. It has been suggested that trauma may increase the risk of psychosis across diagnostic boundaries, through enduring changes in the cortisol feedback loop within these systems (Reference Collip, Myin-Germeys and WichersCollip 2013). Both the ANS and the HPA axis modulate the reactivity of the immune system, thereby implicating a close relationship between the different stress response systems. However, again confounding factors, including medication effects, have rarely been controlled for in studies.
In one recent investigation of drug-naive first-episode psychosis, Reference van Venrooij, Fluitman and Lijmervan Venrooij et al (2012) showed a lower cortisol and adrenocorticotrophic hormone (ACTH) response to a stressful event, with reduced natural killer cell levels, compared with matched controls; the autonomic response remained intact. They suggest that this indicates a potentially different biological stress response already present in medication-naive patients with first-episode psychosis. High levels of circulating pro-inflammatory cytokines have been seen in people with unmedicated first-episode psychosis compared with healthy controls. In patients with a history of childhood trauma and recent life events, these levels are higher still, suggesting that the predisposing and ongoing immune modulatory effects of environmental stressors are not medication related (Reference Miller, Buckley and SeaboltMiller 2011).
Evidence from genetic studies
Clinical experience and many years of research demonstrate the heritability of a risk of schizophrenia, as shown in twin, adoption and family studies (Reference Aberg, Liu and BukszárAberg 2013). Yet with mapping of the human genome, this knowledge has not readily translated into replicable findings for schizophrenia as an entity with clearly defined borders and there is overlap with affective disorders and autism. Craddock and others propose complex interaction of genetic, epigenetic and environmental influences that convey risk for major mental illness, not reflected in our current nosological disease boundaries (Reference Craddock and OwenCraddock 2010, Reference Craddock and Sklar2013).
Genome-wide association studies (GWAS), whereby the data of large numbers of patients can be employed to identify, for example, single nucleotide polymorphisms (SNPs), are used to explore individual susceptibility genes in complex disorders. † These studies clearly indicate immunological pathways in psychosis. Robust findings demonstrate an association of schizophrenia with markers in the genetic coding region for the major histocompatibility complex (MHC), the cell surface macromolecule complex mediating interaction between different white blood cells (Reference Stefansson, Ophoff and SteinbergStefansson 2009). These results suggest that prenatal exposure to infection can modify MHC function, via gene expression, conferring greater susceptibility to schizophrenia post-puberty (Reference Kinney, Hintz and ShearerKinney 2010). Significant associations have been found between alleles at several loci on the MHC regions in chromosome 6 and increased risk for schizophrenia in a meta-analysis of extensive data from GWAS studies of 12 000 patients with schizophrenia (Reference Stefansson, Rujescu and CichonStefansson 2008). A number of autoimmune diseases are also associated with alleles at some of these same loci. In addition, a study looking at 22 278 genes found significant differential expression in 200 genes implicated in schizophrenia, the most significant of which were 20 that have effect in seven biological processes related to protein processing and maturation, innate immune response and the acute inflammatory response (Reference Sainz, Mata and BarreraSainz 2012).
Thus, many studies of recent years show that pathways relating to inflammation and immunity are clearly implicated in schizophrenia (Reference Gardiner, Cairns and LiuGardiner 2013).
Conclusion and clinical implications
Major advances in our understanding of the pathophysiology of schizophrenia suggest immune dysfunction as a common finding. The potential for translation into improved treatments may be within our grasp. Initial evidence from randomised controlled trials of immunomodulatory drugs such as cyclooxygenase-2 (COX-2) inhibitors has shown promising results (Reference Akhondzadeh, Tabatabaee and AminiAkhondzadeh 2007; Reference Müller, Krause and DehningMüller 2010b). There is clear potential for advances using other neuroprotective treatments. Minocycline, an anti-inflammatory neuroprotective antibiotic, is currently being investigated and early results are promising, particularly with regard to negative symptoms (Reference Chaudhry, Hallak and HusainChaudhry 2012).
Our understanding of both the nosological boundaries and the ‘end phenotypes’ of psychotic disorders continues to advance. As presently defined, schizophrenia is a heterogeneous condition, likely to have numerous pathological pathways (Reference Silveira, Marques-Teixeira and De Bastos-LeiteSilveira 2012). However, immune dysfunction is implicated in many current studies of its pathogenesis; thus, the concept of schizophrenia as a ‘non-organic psychosis’ is significantly challenged. Together with our increasing knowledge of the complexities of the cellular and humoral immune system and its genetic coding, this area will continue to provide major advances for our understanding of neurobiological processes in psychotic illness (Reference Falkai and MöllerFalkai 2012). Several factors still need to be taken into consideration, such as precise phenotypic definition of cell subsets, sampling of patients in different stages of disease progression, smoking status, weight, ethnicity and perhaps most importantly exposure to antipsychotic medications. Further study of antipsychotic-naive first-episode psychosis will provide the ideal sample in which to explore these advances in knowledge.
The investigation of immune dysfunction in psychosis offers the greatest potential for advancing our understanding of schizophrenia in the 21st century. Intimate interconnections are apparent between the brain and the immune system, present as a legacy of our evolutionary past and running unchecked in the modern world. The possible consequence of the resulting excessive inflammation is major multiple illnesses affecting the brain, not recognised by current nosological boundaries (Reference Raison and MillerRaison 2013) but including within this spectrum what we currently recognise as schizophrenia. As with the common knowledge and teaching of neurotransmitter actions, structural brain changes and familial risks in psychosis, it is important that all psychiatrists, clinical and academic, remain up to date with understanding of the immune system and advances pertaining to psychosis therein. Meanwhile, increased awareness and clinical implications, such as routine screening of all patients with psychosis for autoimmune encephalitis, and the investigation of novel treatments, need to advance at a similarly brisk pace.
MCQs
Select the single best option for each question stem
-
1 As regards cytokines:
-
a they may be affected by antipsychotic medication
-
b they are components only of the innate immune system
-
c they cannot cross the blood–brain barrier
-
d they are high molecular weight proteins
-
e cytokine imbalance is not implicated in schizophrenia.
-
-
2 As regards microglia:
-
a they are mononuclear phagocytes of the brain
-
b they play a major role in brain development
-
c they are responsible for synaptogenesis
-
d they are responsible for synaptic pruning
-
e all of the above are true.
-
-
3 Cortisol:
-
a acts to stimulate the immune system
-
b is a peptide hormone belonging to the glucocorticoid groups
-
c redistributes glucose to regions of the body required for a fight-or-flight response
-
d is underproduced during chronic stress
-
e singularity modulates the reactivity of the immune system.
-
-
4 Currently tested autoantibodies in the limbic system include:
-
a NMDA receptor
-
b GABA-BR1
-
c GABA-BR2
-
d VGKC
-
e all of the above.
-
-
5 Evidence for immune dysfunction in schizophrenia:
-
a includes an elevated risk of schizophrenia in persons who are born in the summer
-
b was first proposed in 1990s
-
c does not include the microglial hypothesis
-
d includes a proposed imbalance between Th1 and Th2 arms
-
e is of minor importance.
-
MCQ answers
| 1 | a | 2 | e | 3 | c | 4 | e | 5 | d |



eLetters
No eLetters have been published for this article.