

Most of the excitatory neurotransmission in the central nervous system (CNS) is mediated by the endogenous excitatory amino acids (EAAs) glutamate, aspartate and homocysteine. Most of the endogenous inhibitory neurotransmission is mediated by gamma-aminobutyric acid (GABA). EAAs modulate the firing of almost all neurons in the CNS, as excitatory neurotransmission can result in both neuronal inhibition and excitation. The glutamate system is the best characterised of the EAA systems (Box 1).
Box 1 Facts about glutamate
-
It is the primary excitatory neurotransmitter in the human central nervous system
-
It acts on the N-methyl-d-aspartate (NMDA) receptors and the non-NMDA amino-3-hydroxy-5-methyl-4-isoxazole propionic acid and kainate receptors, which are all ion channels
-
It also acts on types I, II and III metabotropic receptors, which are G-protein coupled
Glutamate is widely distributed in the CNS, and a large body of evidence indicates its involvement not just in fast synaptic transmission but also in plasticity and higher cognitive functions. Glutamate can also induce neurotoxicity, and it has therefore been implicated as a potential contributor to the pathogenesis of several CNS neurodegenerative disorders, for example Alzheimer's disease.
Glutamatergic receptors
The post-synaptic effects of glutamate are mediated via several receptor subtypes. Different combinations of these subtypes determine the specific functional capability of individual synapses and neurons. Since many combinations of subunits are possible for each type of glutamatergic receptor the system is extremely complex. There are four types of glutamatergic receptor (see Box 1), each with unique regional and synaptic distributions. They include both the ionotropic and metabotropic receptor families. Ionotropic receptors allow the passage of charged ions into the neuron, for example chloride (Cl–) and calcium (Ca2+). Metabotropic receptors function via chemical second messengers within the neuron. Glutamatergic receptors are grouped according to similarities in amino acid sequence and pharmacodynamic properties such as affinity for glutamate and threshold for channel opening.
Ionotropic glutamatergic receptors
Ionotropic glutamatergic receptors open cation-permeable channels to mediate sodium (Na+), potassium (K+) or calcium (Ca2+) ion flow. There are three families of ionotropic receptors: the N-methyl-d-aspartate (NMDA), the amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and the kainate receptors.
The AMPA and the kainate receptors are collectively termed ‘non-NMDA’ receptors and appear to control conductance of Na+ and K+ through channels that exhibit rapid kinetics. AMPA receptors are predominantly post-synaptic receptors, widely distributed in the cortex and ventral striatum and in temporal lobe structures such as the hippocampus and amygdala, with lower levels in the thalamus. Of the three ionotropic receptors in the CNS, AMPA receptors occur at the greatest density. They include GluR1, GluR2, GluR3 and GluR4. The agonists acting at AMPA receptors are AMPA and amino-3-hydroxy-5-tert-butyl-4-isoxazole propionic acid (ATPA). They mediate most fast excitatory transmissions in the brain. Kainate receptors are predominantly presynaptic and appear to regulate glutamate release. They include GluR5, GluR6, GluR7, GluR KA-1 and GluR KA-2. They appear to be distributed mainly in the hippocampus and the infragranular layers of the cortex. Kainate receptor agonists include kainate and domoic acid.
The most structurally complex glutamatergic receptor is the NMDA receptor. This is the glutamate receptor most often implicated in neuropsychiatric disorders. It is an ion channel made up of different and variably assembled protein isoforms. It is the only ionotropic receptor to control Ca2+ conductance in addition to the conductance of Na+ and K+. When the channel is activated there is an influx of Na+ and Ca2+ ions and an efflux of K+ ions. Binding of magnesium (Mg2+) ions to sites within the channel prevents Ca2+ influx. Activation of the channel can occur only if there is simultaneous glutamate and glycine binding and partial depolarisation of the membrane potential. Glycine is an ‘obligate co-agonist’ for glutamate, i.e. glutamate cannot act on the NMDA receptor in the absence of glycine. The simultaneous binding of the two transmitters and partial depolarisation permits Mg2+ displacement and channel opening.
The NMDA receptor is found predominantly post-synaptically and seems to be concentrated primarily in the limbic system, co-localised with AMPA receptors. The voltage dependence of NMDA receptors has the effect of enhancing the depolarisation initiated by non-NMDA receptor channels. Ca2+ can subsequently act as a second messenger and initiate a wide range of intracellular responses that underlie a number of complex neurophysiological phenomena. NMDA receptor activity is under the influence of several factors, and the receptors have several different regulatory sites of interest. NMDA receptors include NMDA R1 (isoforms A–G) and NMDA R2 (isoforms A–D). The ion channel regulated by these receptors is blocked by phencyclidine (PCP), ketamine and the NMDA analogue MK–801. Potent direct-acting agonists at the NMDA receptors are NMDA and glutamate.
Metabotropic glutamatergic receptors
Metabotropic glutamatergic receptors are activated via G-proteins rather than via cation channels. They have seven transmembrane domains and, being G-protein coupled, they are similar to most dopamine, serotonin and noradrenaline receptors, activating phospholipase C or inhibiting adenylate cyclase. They are divided into three groups: type I (mGluR1 and 5), type II (mGluR2 and 3) and type III (mGluR4, 6, 7 and 8). Each group appears to have specific functions (Reference Meador-Woodruff and HealyMeador-Woodruff & Healy, 2000). Type I are post-synaptic, occurring in hippocampal, amygdala and thalamic neurons and, to a lesser extent, in the cortex and ventral striatum. Types II and III are presynaptic modulators of glutamate release. Within type II, mGluR2 is restricted to the cortex and dentate gyrus, whereas mGluR3 appears to be the only metabotropic subtype expressed on glia.
Glutamate and schizophrenia
The dopamine hypothesis remains the most accepted model for schizophrenia and is supported by persuasive evidence, in particular dopamine mediated amphetamine-induced psychosis and the close relationship between clinical response and antipsychotic-induced dopamine D2 receptor blockade. Neuroimaging studies have shown, however, that a degree of D2 occupancy could be necessary but not sufficient to account for the beneficial effects of antipsychotic drugs. Furthermore, non-responders to medication still show high levels of D2 blockade (Reference Pilowsky, Costa and EllPilowsky et al, 1993). It has subsequently been suggested (Reference Laruelle, Abi-Dargham and van DyckLaruelle et al, 1996) that abnormalities could also reside in the control of dopamine release rather than in absolute dopamine receptor alterations. Thus, interest has been focusing on systems modulating or acting on dopamine neurons.
Early indirect evidence that glutamate may be involved in schizophrenia came from studies using 18F-fluorodeoxyglucose positron emission tomography (PET) scanning comparing floridly psychotic drug-free patients with normal controls. Findings suggested that the areas with the highest concentration of glutamate receptors – the anterior cingulate cortex, the medial surface of the frontal lobe and the hippocampal formation – seem to be abnormal or to function abnormally in positive-symptom aspects of schizophrenia (Tamminga et al, 1992). More direct evidence has been provided from pharmacological studies with NMDA receptor antagonists such as PCP and ketamine.
Phencyclidine
Phencyclidine (‘angel dust’) is the prototypic non-competitive NMDA-receptor antagonist, inducing the characteristic behavioural changes described as early as 1959. To a great extent its behavioural actions approximate some of the signs and symptoms of schizophrenic psychosis, perhaps even more faithfully than the amphetamines (Reference Javitt and ZukinJavitt & Zukin, 1991). Unlike other psychotomimetic drugs, PCP produces a range of classic psychotic phenomena in normal individuals in a dose-dependent manner. These include delusions, thought disorder and hallucinations. Early data suggested that PCP might exacerbate existing symptoms in schizophrenia, rather than producing new symptoms arising from its psychotomimetic action. The glutamatergic hypothesis was taken further by Anis et al (1983), who were the first to report that PCP and its congeners (e.g. ketamine) blocked the action of NMDA on ion flow through the NMDA-sensitive glutamate receptor in the brain. A PCP receptor (the ‘PCP site’) was soon identified and characterised, and its location within the NMDA-gated glutamate ionophore was further characterised. It also became evident that PCP receptor stimulation antagonised NMDA receptor function at relatively low concentrations, suggesting that the low-dose psychotomimetic actions of PCP were associated with the high-potency blockade activity of PCP, and could therefore be discrimated from its broad pharmacological profile.
Reports of psychotomimetic actions of other competitive NMDA antagonists suggest that any blockade of this ion channel will cause psychotomimetic side-effects (Reference Grotta, Clark and CoullGrotta et al, 1995). These observations have supported the hypothesis that any drug or disease that reduces glutamatergic transmission at the NMDA site could be associated with psychosis. Conversely, both the conventional antipsychotic haloperidol and the atypical clozapine mediate gene expression (c-fos) via intracellular regulation of NMDA receptors in the striatum. The cAMP pathway thus activated leads to phosphorylation of the R1 subtype of the NMDA receptor at 897Ser (Reference Leveque, Macias and RajadhyaskshaLeveque et al, 2000). It has therefore been hypothesised that antipsychotic drugs have the ability to modulate NMDA receptor function and that this facilitation of NMDA activity is necessary for antipsychotic-drug-mediated gene expression and may contribute to both the therapeutic benefits and the side-effects of antipsychotic treatment.
Ketamine
Ethical considerations have prohibited the full exploration of PCP effects in humans. However, its derivative, ketamine, provides a safer experimental tool as it has been widely used as an anaesthetic agent with an established record of safety in healthy humans. Indeed, it has been suggested that sub-anaesthetic doses of ketamine produce in healthy subjects three clusters of symptoms that have been described in individuals with schizophrenia: the positive, negative and disorganisation symptoms (Reference Andreasen, Arndt and AlligerAndreasen et al, 1995). The extent to which ketamine produces these effects is related to the dose and rate of infusion (Reference Newcomer, Farber and Jevtovic-TodorovicNewcomer et al, 1999). In some subjects, the positive symptoms produced by ketamine are indistinguishable from symptoms seen in patients with schizophrenia (Krystal et al, 1999a), but there are also differences. Auditory hallucinations, for example, are produced infrequently by ketamine, and perceptual alterations closely resembling dissociative states are a prominent behavioural effect of ketamine. In addition, it has been suggested that the degree to which NMDA antagonists produce symptoms within a given sensory domain is related to the extent of the environmental stimulation within that domain (Krystal et al, 1999b). Thus, dose, pharmacokinetics and environmental manipulations may influence the degree to which ketamine effects seem similar to the positive symptoms of schizophrenia. Interpretation of the negative symptoms produced by ketamine seems to be confounded by its sedative effects, but it has been shown to produce more negative symptoms than lorazepam and haloperidol at similarly sedating doses. In addition, ketamine has been shown to produce bradykinesia in healthy human subjects. It also seems to induce thought disorder that is essentially indistinguishable from thought disorder in subjects with schizophrenia. Furthermore, ketamine has also been shown to impair performance on several tests of cognition (Krystal et al, 1999b), including encoding deficits in memory tests (Reference Krystal, Karper and SeibylKrystal et al, 1994; Reference Newcomer, Farber and Jevtovic-TodorovicNewcomer et al, 1999).
Another interesting aspect of the PCP/ketamine model of schizophrenia is that children are reported to be less sensitive to the psychotomimetic effects of ketamine than are adults (Reference Goff and WineGoff & Wine, 1977). As schizophrenia affects individuals in adolescence or early adulthood and is very uncommon among children, this fact adds weight to the involvement of glutamatergic deficits in the neurobiology of schizophrenia.
A glutamatergic deficiency model of schizophrenia
The mechanisms underlying hyperdopaminergic function in schizophrenia are unclear, but may involve cortical glutamatergic projections to dopamine neurons in the midbrain (Reference Carlsson, Hansson and WatersCarlsson et al, 1997). Dopamine neurons, like other monoaminergic brainstem neurons, seem to be controlled either directly by corticofugal glutamatergic neurons, which act as accelerators, or indirectly via GABAergic interneurons, which act as brakes (Fig. 1.). Low glutamate concentration may then cause an increase or decrease in dopamine function, depending on whether the effect on the brake or the accelerator predominates. Under normal circumstances the brake appears to be in approximate equilibrium with the accelerator, perhaps with the brake exerting its effect slightly more. Thus, a reduced glutamate function, for example as induced by ketamine, may cause some elevation of dopamine release. If, however, dopamine release is massively enhanced, for example by amphetamine, a negative feedback regulatory circuit appears to be activated, leading to augmentation of the brake. This has been demonstrated in humans by single-photon emission computed tomography studies that have shown that co-treatment with the NMDA antagonist ketamine causes enhancement of amphetamine-induced dopamine release (Reference Kegeles, Abi-Dargham and Zea-PonceKegeles et al, 2000). This is a clinically relevant observation, as it offers a possible explanation for the enhanced amphetamine-induced dopamine release in patients with schizophrenia (Carlsson et al, 2001). This hyperdopaminergia could be secondary to a glutamate deficiency at the level of the NMDA receptor, which leads to a weakened negative feedback mechanism, possibly via the thalamus and the striatum.
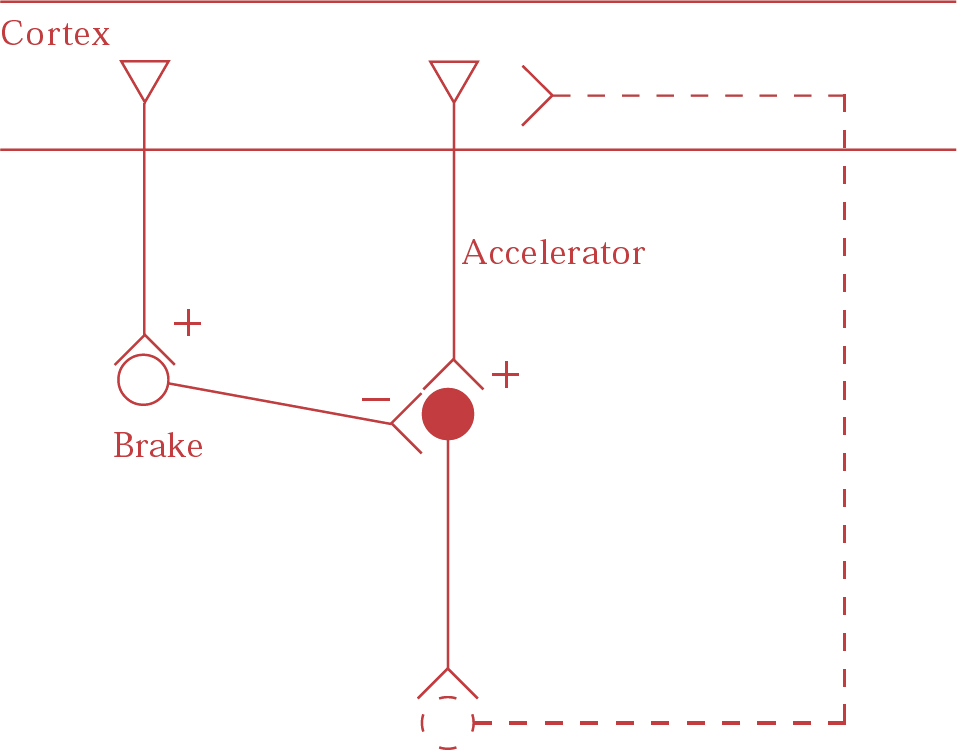
Fig. 1 Cortical glutamate regulates brainstem monoaminergic neurons in two possible ways: (a) directly, by means of glutamatergic fibres projecting to brainstem neurons (accelerator); and (b) indirectly by fibres projecting to the glutamatergic/gabaergic pathway from cortex to brainstem (brake). Feedback loops probably exist, probably via the thalamus and the striatum. (From Reference Carlsson, Hansson and WatersCarlsson et al, 1999.)
Serotonin–glutamate interactions
Treatment of experimental animals with NMDA antagonists alone has yielded variable results, and different parts of the dopaminergic system have been found to respond differentially to treatment with the NMDA analogue MK–801. It is thus evident that a mechanism other than increased dopamine release accounts for at least part of the psychostimulant and psychotogenic action of NMDA antagonists. The serotonin (5-hydroxytryptamine or 5-HT) hypothesis of schizophrenia arose from early studies on interactions between the hallucinogenic drug d-lysergic acid diethylamide (LSD) and serotonin in peripheral systems. Further studies have shown that the two major classes of psychedelic hallucinogen, the indoleamines (e.g. LSD) and the phenethylamines (e.g. mescaline), produce their central effects through a common action on 5-HT2A receptors. In each case, these drugs have been found to enhance glutamatergic transmission. In the prefrontal cortex, 5-HT2A receptor stimulation increases the release of glutamate. This effect is blocked by inhibitory types II and III metabotropic glutamate agonists acting presynaptically and by an AMPA or kainate glutamate antagonist, acting post-synaptically at non-NMDA glutamate receptors (Reference Aghajanian and MarekAghajanian & Marek, 2000).
Hypo- or hyperglutamatergic states and psychosis
In the PCP (or ketamine) model of psychosis, it appears that decreased function at the NMDA receptors is the drive behind psychosis, perhaps via dopaminergic mechanisms. In the LSD model, there appears to be an increase in glutamatergic function. These seemingly paradoxical findings may be reconciled by considering the differences between the various receptors on which glutamate may act. For example, the blocking of NMDA receptors by PCP or ketamine may lead to a compensatory and net increase in glutamatergic function and hence to an increase in the release of glutamate acting on non-NMDA receptors (AMPA and kainate) (Reference Moghaddam, Adams and VermaMoghaddam et al, 1997). Furthermore, it has been hypothesised that a decrease in the function of NMDA receptors removes the excitatory drive to inhibitory GABAergic neurons. These GABAergic neurons, thus affected, also regulate non-NMDA excitatory neurons acting on areas such as the frontal cortex and the limbic regions, decreasing inhibitory control, increasing firing and producing psychotic symptoms (Reference Farber, Hanslick and KirbyFarber et al, 1998). Thus, LSD might produce psychosis by increasing glutamatergic function at non-NMDA receptors, whereas PCP and ketamine might produce it by reducing glutamatergic function at NMDA receptors (Box 2). It is interesting to note that hallucinations, which are the prime symptom in LSD-type psychoses, are rarely seen in PCP- or ketamine-induced psychosis and that negative symptoms are rarely seen in LSD-related psychoses. This might reflect differences in the contribution of NMDA and non-NMDA receptor function to the complex syndrome of schizophrenia.
Box 2 Glutamate and schizophrenia
-
Psychotic symptoms can be produced by decreased glutamatergic function at NMDA receptors
-
Psychotic symptoms can also be produced by increased glutamatergic function at AMPA and kainate receptors
-
Drugs acting at the glycine site on the NMDA receptor may be useful in the treatment of some symptoms in schizophrenia
Treatment implications
There is clearly need to explore new treatment approaches in schizophrenia, as even the best therapeutic responses obtained with existing typical or atypical antipsychotic drugs are often delayed and not fully restorative (Reference TammingaTamminga, 1998). A possible reason for this lack of full efficacy may be that the primary site of pathology for schizophrenia may lie ‘upstream’ or ‘downstream’ of the receptors that are targeted by the currently available drugs (i.e. the D2-like receptors, the 5-HT2A receptor, etc.). If hyperglutamatergic states play a role in the pathogenesis of schizophrenia, as they do in LSD-related psychotomimetic drug models, then treatments that limit or suppress glutamate release may be therapeutic or prophylactic in this disease. As glutamate is the main excitatory neurotransmitter in the CNS a generalised block of glutamatergic transmission would be potentially dangerous. An alternative approach is to target the hypothesised underlying hypoactivity of NMDA receptors directly or indirectly. By acting at the NMDA subtype of the glutamate receptor, these putative treatments enhance the activity of NMDA receptors, which could correct underlying hypoactivity. Directly acting agents have, however, been unsuccessful, in part because of adverse effects such as memory impairment and neurotoxicity, potentially including seizures and catatonic reactions (reviewed in Reference Bressan and PilowskyBressan & Pilowsky, 2000). Since glycine-related compounds act indirectly as obligatory co-agonists increasing the frequency of the NMDA-gated channel openings, medications that act at the glycine site have subsequently been tested.
Treatment trials of glycine and its analogues
Three double-blind, placebo-controlled trials of glycinergic agents used to augment conventional antipsychotic treatment have been published. Tsai et al (1998) conducted a trial of d-serine on 31 patients with schizophrenia and found significant improvements in positive, negative and cognitive symptoms. Goff et al (1999) performed a trial using d-cycloserine on 47 patients and found a significant reduction in negative symptoms, but no improvement in positive or cognitive symptoms. Heresco-Levy et al (1999) evaluated 21 treatment-resistant patients with schizophrenia in a crossover treatment trial of glycine and found a significant reduction in negative symptoms. These findings of improvement of schizophrenic symptoms with glycinergic agents support the NMDA receptor hypofunction hypothesis in schizophrenia and suggest novel approaches for the pharmacotherapy of the disorder, particularly of its negative symptoms.
New targets for antipsychotics based on psychotomimetic drug models
Another approach to the pharmacological manipulation of glutamatergic neurotransmission would be to explore 5-HT2A-receptor-mediated glutamate release. NMDA receptor antagonists appear to stimulate 5-HT turnover and release more consistently than dopaminergic activity. The selective 5-HT2A antagonist R(+)—(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidinemethanol (M100907, also known as MDL 100,907) has a striking effect on the behavioural stimulation induced by NMDA receptor antagonism (Reference Martin, Waters and SchmidtMartin et al, 1998). The non-dopaminergic profile of M100907 is very different from that of other antipsychotic agents and this seemed to have important therapeutic implications, especially since post-mortem data suggest a presynaptic hyperserotonergia in individuals with paranoid schizophrenia (Reference Hansson, Waters and WinbladHansson et al, 1994). Despite favourable results in open label phase II trials, M100907 is no longer being developed.
Although M100907 was apparently not successful, in support of its mechanism of action it is worth noting that the psychostimulation caused by PCP does appear to be relatively independent of dopamine release. This psychostimulation can be nearly abolished by LY354740, a type II metabotropic glutamate receptor agonist, despite the fact that this agonist leaves the enhanced dopamine release unchanged (Moghaddam & Reference Moghaddam and AdamsAdams, 1998). Both LY354740 and the type II/III metabotropic agonist (1S, 3S)-ACPD, which reduce the release of glutamate by acting on presynaptic inhibitory autoreceptors, are able to block excitation induced by 5-HT2A receptors in vitro (Reference Marek, Wright and SchoeppMarek et al, 2000). Similarly, in rats LY354740 has been shown in vivo to improve some cognitive deficits produced by the NMDA antagonist PCP (Moghaddam & Reference Moghaddam and AdamsAdams, 1998). These results suggest that metabotropic agonists may be useful in normalising excesses in glutamate release, regardless of their cause. The availability of orally active metabotropic glutamate receptor agonists makes it feasible to test the hypothesis that excessive glutamate release, particularly in such critical cortical regions as the prefrontal cortex, play a role in the positive and/or negative symptoms of schizophrenia.
Lamotrigine attenuation of ketamine-induced effects in humans
In addition to metabotropic glutamate receptor agonists and glycine, glutamate release can be decreased by lamotrigine (3,5-diamino-6-[2,3-dichlorophenyl]-1,2,4-triazine), an anticonvulsant that stabilises neuronal membranes and attenuates cortical glutamate release via inhibition of use-dependent Na+ channels and P- and N-type calcium channels, and via its effects on K+ channels. Anand et al (2000) have recently shown that lamotrigine reduces the neuropsychiatric effects induced in healthy humans by sub-anaesthetic doses of ketamine. However, further studies are needed because lamotrigine affects other neurotransmitter systems (e.g., 5-HT, GABA and dopamine) and it is unclear whether these effects are direct or secondary to the effects on glutamate release.
Glutamate and mood disorders
In the study by Anand et al (2000), lamotrigine potentiated the mood-elevating effects of the NMDA antagonist ketamine, thus suggesting that mood elevation may be associated with a decrease in excitatory amino acid transmission. Indeed, lamotrigine has been reported to be useful in the treatment of bipolar depression (Reference Calabrese, Bowden and SachsCalabrese et al, 1999). NMDA-receptor-modulating drugs may also be important in the treatment of depression. A growing body of preclinical research suggests that the NMDA glutamate receptors are implicated in the pathophysiology of major depression and the mechanism of action of antidepressants. In many (but not all) studies, NMDA receptor antagonists have been shown to be effective in animal models of depression and models that predict antidepressant activity. Antidepressant administration has also been shown to affect NMDA receptor function and receptor binding profiles. Indeed, chronic administration of 16 out of 17 antidepressant medications consistently reduced the potency of glycine's action at specific glycine sites (Reference Paul, Nowak and LayerPaul et al, 1994). Additional evidence suggests a transcriptional mechanism for this phenomenon, as repeated administration of antidepressants has been shown to alter regional expression of mRNA that encodes multiple NMDA receptor subunits (Reference Boyer, Skolnick and FossomBoyer et al, 1998). Data from a double-blind placebo-controlled trial to assess the treatment effects of a single dose of ketamine in a small number (n = 7) of subjects with major depression were consistent with reports on the use of NMDA receptor antagonists in animal models of depression (Reference Berman, Cappiello and AnandBerman et al, 2000). It was shown that depressive symptoms significantly improved within 72 h of ketamine, but not of placebo, infusion. These findings suggest a potential benefit from further exploration of NMDA antagonists as antidepressant agents, but clinical applicability of this strategy may be limited by the psychotomimetic effects and the potential for misuse of many of these agents. NMDA receptor antagonists without psychotomimetic properties in humans (e.g. memantine), however, merit testing for antidepressant activity.
Glutamate and anxiety disorders
The role of the 5-HT system in anxiety disorders is well established, and chronic administration of the selective serotonin reuptake inhibitors (SSRIs), which are broad-spectrum anti-anxiety medications, produces a net increase in 5-HT neurotransmission, thus augmenting the normally adaptive central 5-HT system response to stress. In particular, a 5-HT2A receptor sensitivity has been shown across anxiety disorders. It should be remembered that it is through their action on precisely these receptors that psychedelic psychostimulants such as LSD enhance glutamatergic neurotransmission. Preclinical data have implicated the NMDA glutamatergic receptor in the acquisition of conditioned fear (e.g. fear-potentiated startle; Reference Davis, Rainnie and CassellDavis et al, 1994). Furthermore, abnormal baseline and fear-potentiated startle have been observed in patients with post-traumatic stress disorder, suggesting that disturbed glutamatergic function contributes to human anxiety (Reference Morgan, Grillon and SouthwickMorgan et al, 1995). Moreover, drugs targeting the metabotropic glutamate receptors mGluR2 and mGluR3 have shown anxiolytic potential in preclinical models of fear and anxiety (Reference Schoepp, Jane and MonnSchoepp et al, 1999). Further studies are imperative before any firm conclusions can be drawn about the role of the glutamate system in anxiety, but it promises to be a fruitful target for ongoing neurobiological and treatment studies of anxiety (Krystal et al, 2001) (see Box 3).
Box 3 Glutamate and other psychiatric disorders
-
NMDA receptor hypofunction has been implicated in the genesis of depressive symptoms
-
Some anxiety symptoms may be related to changes in metabotropic glutamate receptor function
-
The glutamatergic system is a possible therapeutic target in the treatment of neurocognitive disorders
Glutamate and disorders of cognition
Cognitive deficits are an area of renewed interest in the treatment of schizophrenia and other neuropsychiatric disorders. Interestingly, lamotrigine has been shown to lead to a decrease in ketamine-induced learning and memory impairment (Reference Anand, Charney and OrenAnand et al, 2000). It has therefore been suggested that reducing glutamate release might be helpful in disorders characterised by neurocognitive deficits, and it has further been hypothesised that glutamate-induced excitotoxic effects may lead to widespread neuronal degeneration such as that seen in Alzheimer's disease (Reference Olney, Wozniac and FarberOlney et al, 1997). Preliminary reports suggest some efficacy of lamotrigine in the treatment of Alzheimer's disease (Reference Tekin, Aykut-Bingol and TanridagTekin et al, 1998), but this requires replication before firm conclusions can be drawn.
Conclusions
Although the glutamatergic system is highly complex, there is increasing evidence for its involvement in a wide variety of symptoms seen in neuropsychiatric disorders and for the clinical potential of glutamatergic agents. It would appear from the available evidence that there are three main strands emerging. First, that hypofunction of the NMDA receptor system might be involved in the core symptoms of schizophrenia, perhaps by increasing activity in some non-NMDA systems and/or by a direct effect on dopaminergic neurotransmission. Second, that hyperfuntion in non-NMDA systems might underlie some of the more florid positive symptoms in psychotic disorders. And third, that metabotropic glutamatergic receptors might play an important role in anxiety disorders.
Much remains to be learned about the possibilities of using this knowledge in the development of novel pharmacological strategies. Early work, reviewed briefly here, seems to suggest that medications that affect glutamatergic function may have a role as augmentors of treatment, as is the case with glycine analogues in schizophrenia, and perhaps as monotherapy in bipolar depression, where lamotrigine might be efficacious.
The involvement of glutamate in the regulation of neural networks suggests that a combination of research techniques including neuroimaging, pharmacological challenge tests, neuropsychology, neurophysiology and pharmacogenetics, among others, will help us to elucidate the role of each part of the glutamatergic system in psychiatric disorders and thus to develop novel but pharmacologically rational treatments.
Multiple choice questions
-
1. In regard to glutamate receptors:
-
a AMPA receptors are G-protein coupled
-
b glycine is an obligate co-agonist at NMDA receptors
-
c type II metabotropic glutamatergic receptors are presynaptic
-
d kainate receptors have a higher density than AMPA receptors in the human brain
-
e the NMDA receptor is a calcium channel.
-
-
2. Psychotomimetic drugs that act directly via the NMDA receptor include:
-
a phencyclidine (PCP)
-
b met-amphetamine
-
c d-lysergic acid diethylamide (LSD)
-
d ketamine
-
e mescaline.
-
-
3. Rate the following statements as true or false:
-
a psychosis may be induced by antagonism of NMDA receptors
-
b hallucinations are associated with stimulation of AMPA and kainate receptors
-
c medications acting directly at the glutamate site on the NMDA receptor are theoretically very safe
-
d d-serine has similar actions to glycine
-
e medications acting at the glycine site on the NMDA receptor may be preferentially helpful for the negative symptoms of schizophrenia.
-
-
4. Lamotrigine:
-
a reduces glutamate release in the cortex
-
b might be useful in the treatment of bipolar depression
-
c enhances the effects of ketamine in healthy volunteers
-
d has pro-convulsant effects similar to those of clozapine
-
e is an effective antipsychotic when used alone.
-
-
5. The following are antagonists at the corresponding receptors:
-
a MK– 801 at NMDA receptors
-
b M100907 at 5-HT1A receptors
-
c met-amphetamine at D2 receptors
-
d ketamine at mGluR1 receptors
-
e LSD at 5-HT2A receptors.
-
MCQ answers

| 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a | F | a | T | a | T | a | T | a | T |
| b | T | b | F | b | T | b | T | b | F |
| c | T | c | F | c | F | c | F | c | F |
| d | F | d | T | d | T | d | F | d | F |
| e | T | e | F | e | T | e | F | e | F |


eLetters
No eLetters have been published for this article.