

Several studies have demonstrated higher rates of cardiovascular death among psychiatric patients, particularly those with schizophrenia, compared with the general population (Reference Waddington, Youssef and KinsellaWaddington et al, 1998). Cardiac risk factors, including smoking, lack of exercise, obesity, substance-misuse and high autonomic arousal during physical restraint, are overrepresented in psychiatric patients. As psychotropic medication is prescribed for this at-risk population, it becomes necessary to consider its cardiovascular effects. Here we focus on antipsychotics and antidepressants, as the cardiac effects of benzodiazepines and other psychotropics are negligible by comparison.
Heart rate
Resting heart rate arising from spontaneous depolarisation of the sinoatrial node is largely determined by vagal parasympathetic tone. Respiratory sinus arrhythmia (heart rate increase) during inspiration and decrease on expiration is a physiological occurrence. It decreases with age, reflects heart rate variability and, like baroreceptor reflex sensitivity, is determined by sympathovagal balance but mainly depends on vagal influences on the sinoatrial node.
Psychotropic drugs possessing antimuscarinic and anticholinergic effects (e.g. all low-potency typical antipsychotics; some atypical antipsychotics, particularly clozapine; most tricyclic antidepressants; non-selective monoamine oxidase inhibitors (MAOIs); and all antiParkinsonian anticholinergics) cause sinus tachycardia which, in otherwise healthy individuals, rarely leads to symptoms and usually remits over time. An exception is clozapine-related myocarditis and cardiomyopathy, presenting with a persisting sinus tachycardia and accompanied by fever, chest pain, palpitations, dyspnoea, ankle oedema or heart failure early on in treatment (Reference Kilian, Kerr and LawrenceKilian et al, 1999) (see below).
In contrast, selective serotonin reuptake inhibitors (SSRIs) produce minor degrees of bradycardia that can usefully reduce cardiac workload and benefit the diseased heart. For example, in one study of elderly patients with depression who also had ischaemic heart disease, fluoxetine 20–60 mg/day induced a 6% asymptomatic decrease in heart rate, whereas nortriptyline, regarded as one of the safer tricyclics in heart disease, increased heart rate by 9%, possibly contributing to more adverse cardiac events (sinus tachycardia, severe angina and ventricular ectopics) (Reference Roose, Glassman and AttiaRoose et al, 1998a ).
Heart rate variability is reduced in medical, psychiatric and psychological conditions such as myocardial infarction, heart failure, heart transplantation, normal ageing, depression, panic disorder, anger, hostility, stress and diabetic autonomic neuropathy, and by the use of full-dose anticholinergic, anticholinergic antipsychotic, and tricyclic antidepressant therapy (Reference Gorman and SloanGorman & Sloan, 2000). Reduced heart rate variability post-myocardial infarction carries a poor prognosis of increased cardiac mortality and sudden death from unopposed sympathetic arrhythmogenic influences (Reference Bigger, Breithardt and CammBigger et al, 1996). Anticholinergic medications such as imipramine reduce heart rate variability further by decreasing vagal tone in depression and panic disorder, but the non-anticholinergic paroxetine has the opposite effect.
Clozapine, a powerful anticholinergic, reduces heart rate variability in patients with schizophrenia (Reference Agelink, Majewski and WurthmannAgelink et al, 2001). Normal heart rate variability, however, can be restored by switching from clozapine to olanzapine, possibly reflecting olanzapine's more modest anticholinergic actions. (Reference Cohen, Loewenthal and MatarCohen et al, 2001).
After myocardial infarction, heart rate variability is maximally, but not fully, recovered within 6–12 months. It is not known whether any recovery from psychotropic-induced heart rate variability reduction occurs or whether this carries the same dire cardiac prognosis as for post-myocardial infarction patients (Reference Bigger, Breithardt and CammBigger et al, 1996).
Strategies for increasing heart rate variability would therefore appear desirable, and these include physical exercise, b-blockade and low-dose anti-cholinergic hyoscine hydrobromide (scopolamine) therapy, which paradoxically increases vagal parasympathetic activity (Reference Bigger, Breithardt and CammBigger et al, 1996). This paradoxical finding might explain the poorer cardiac outcome in patients with chronic schizophrenia who are not co-treated with anticholinergic medication (Reference Waddington, Youssef and KinsellaWaddington et al, 1998).
Conduction
Impulses arising in the sinoatrial node spread across the atria to the atrioventricular node, bundle of His, right and left bundle branches and the distal portions of the Purkinje fibres. The PR interval measures the atrioventricular conduction time and the QRS measures conduction from the atrioventricular node through the bundle of His, bundle branches, Purkinje fibres and ventricular myocardium. The QRS is further subdivided into two intervals that can be measured by bundle of His electrocardiography. The first interval (normal range 50–120 ms) reflects the time taken for an impulse to traverse the atrioventricular node, and the second (normal range 35–55 ms) is the time taken to travel the bundle of His, bundle branches and Purkinje fibres (Reference Burrows, Vohra and HuntBurrows et al, 1976).
Tricyclic antidepressants delay cardiac conduction mainly by prolonging the second part of the QRS interval without significantly affecting the first. The effect is dose-dependent, occurring at both therapeutic and toxic doses, particularly with amitriptyline, imipramine and nortriptyline, but not doxepin (Reference Burrows, Vohra and HuntBurrows et al, 1976). It also probably occurs with dothiepin, but to a lesser degree with lofepra-mine; the perceived difference for doxepin, however, may simply reflect lower plasma levels. This effect is unlikely to be of clinical relevance in the otherwise normal heart. However, it might precipitate complete heart block in the presence of pre-existing conduction delay such as bundle branch block or second and higher degrees of heart (atrioventricular) block. In addition, emergent ventricular pacemakers, which would normally produce an idioventricular rhythm, may be suppressed by the antiarrhythmic action of the tricyclics, thus causing ventricular asystole. Tricyclic antidepressants are more likely to produce higher degrees of heart block in patients with pre-existing bundle branch block (QRS >12 ms) rather than first-degree block (PR >200 ms) (Reference Roose, Glassman and GiardinaRoose et al, 1987) and they should therefore be avoided in all cases of conduction disease beyond first-degree block. None the less, tricyclics are best avoided in patients with any degree of pre-existing conduction delay, because safer alternatives are available with SSRIs.
The cardiotoxicity and mortality from overdose of tricyclic antidepressants is well established. Toxicity arises from sodium (Na+) ion-channel blockade, known as Type 1 antiarrhythmic action. This reduces inward Na+ depolarising current at the beginning of the action potential, leading to conduction delay, bradycardia, atrioventricular block, bundle branch block and monomorphic ventricular tachycardia.
Brugada syndrome (Box 1), another cardiac Na+ channelopathy, first described in 1992, causes idiopathic ventricular fibrillation in structurally normal hearts and sudden unexpected death in about 30% of untreated cases. It is associated with mutation of the SCN5A gene encoding cardiac Na+ channels, causing a reduction of inward Na+ depolarising currents. Electrocardiogram (ECG) features characteristically include right bundle branch block and S–T segment elevation in chest leads V1–V3, but normal QT interval (Fig. 1). Overdose of a tricyclic antidepressant may present with these features. Brugada syndrome is a leading cause of death in Thai males, typically during sleep; 40% of those with the syndrome have a family history of sudden death. It has an estimated prevalence in certain populations of up to 3%.
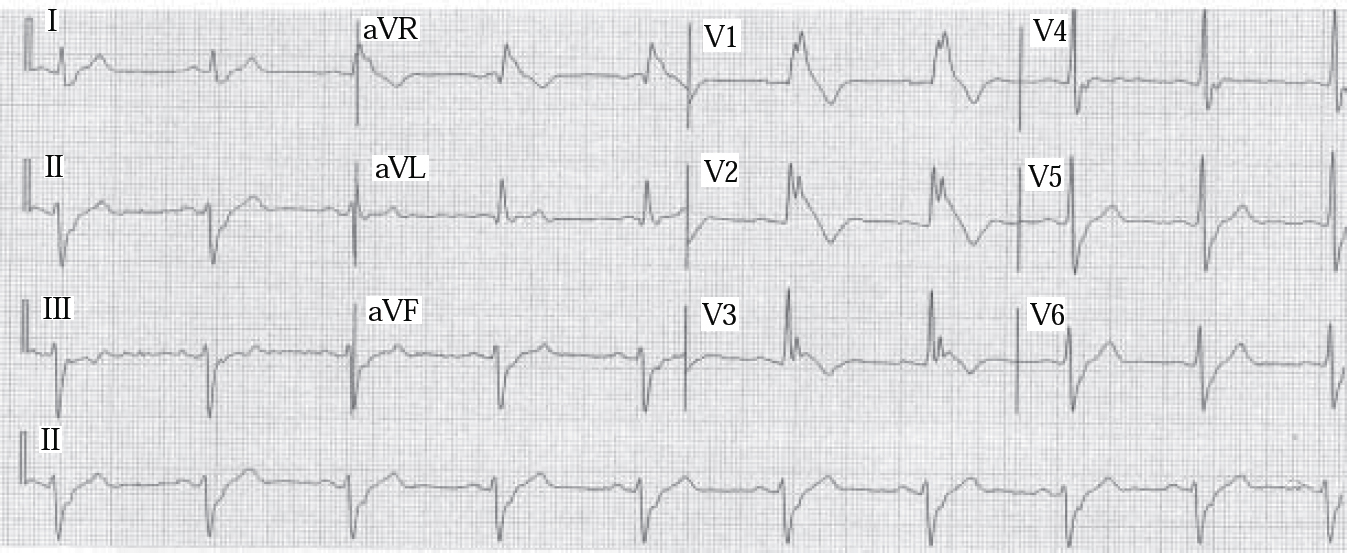
Fig. 1 Brugada syndrome: electrocardiogram demonstrates typical findings of right bundle branch block and elevated S–T segment leads V1–V3. Rhythm: 25 mm/s. (reproduced with permission from Reference Alings and WildeAlings & Wild, 1999).
Box 1 Brugada syndrome
Cause of ventricular fibrillation and sudden unexpected death
Common in young Thai males
40% of patients have a family history of sudden death
Structurally normal heart
Characteristic ECG changes of right bundle branch block and S–T segment elevation leads V1–V3
Normal QT interval
Associated with mutation of SCN5A gene, blocking Na+ channels
Associated with tricyclic antidepressant/neuroleptic toxicity
May present with sudden death/syncope/seizures
Similar appearances have been reported in overdose with tricyclic antidepressants such as amitriptyline and with antipsychotics, but no case was complicated by ventricular arrhythmia and the characteristic ECG pattern disappeared within days. It appears that Na+ channel blockers such as the tricyclics and antipsychotics might provoke Brugada syndrome in otherwise asymptomatic but genetically predisposed individuals.
Type 1 action is also shared with the antiarrhythmic agents encainide, flecainide and moracizine, a phenothiazine analogue. The first placebo-controlled trial of ventricular premature contraction suppression in post-myocardial infarction patients was prematurely stopped because of increased mortality in the encainide and flecainide groups (Cardiac Arrhythmia Suppression Trial II Investigators, 1992). The second trial, run by the same team and reported in the same paper, continued with moracizine v. placebo alone, but this was also abandoned early because of the significant increase in mortality and cardiac arrest in the moracizine group (3% v. 0.4%). Consequently, tricyclic antidepressants and other agents possessing type 1 actions should be avoided, where possible, in psychiatric patients with ischaemic heart disease.
The available SSRIs, in contrast, do not tend to prolong cardiac conduction, with the possible exception of minor conduction delays and QTc prolongation (see below) in citalopram overdose. Even with amounts more than 100 times the therapeutic dose of citalopram, however, the effects on cardiac conduction are few and non-cardiogenic seizures, rather than adverse cardiac events, are the more likely (Reference GlassmanGlassman, 1998). In cardiac patients with pre-existing bundle branch block (QRS >120 ms), therapeutic doses of fluoxetine did not prolong conduction (Reference Roose, Glassman and AttiaRoose et al, 1998a ); neither did paroxetine or sertraline in similar cardiac settings (Reference Roose, Laghrissi-Thode and KennedyRoose et al, 1998b ; Reference Glassman, O'Connor and CaliffGlassman et al, 2002).
Considering their established benefits on heart rate, heart rate variability, cardiac rhythm and platelet function (see below), SSRIs seem a treatment of choice for cardiac patients with anxious depression.
Although antipsychotic medication shares the type 1 antiarrhythmic actions of the tricyclics, by contrast, there is little published evidence that this is clinically significant in therapeutic doses, although in overdose, thioridazine in particular induces greater conduction delay and QRS widening than other typical antipsychotics. None the less, as for tricyclics, it would seem prudent to exercise caution when using such agents in psychiatric patients with a history of ischaemic heart disease or family history of sudden death, especially in view of their proarrhythmic QT-prolonging effects (see below).
Rhythm
Psychotropic impact on cardiac rhythm focuses mainly on the propensity of antipsychotics to affect ventricular repolarisation. Such effects include prolongation of the QT interval (a measure of ventricular repolarisation), a known risk factor for torsade de pointes (‘twisting of the points’), a polymorphic ventricular tachycardia classically associated with long QT interval (Fig. 2).
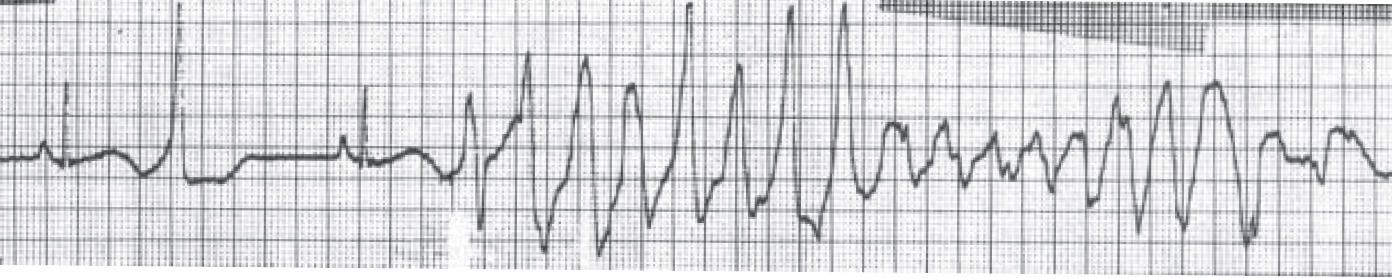
Fig. 2 A short–long–short sequence of beats followed by an episode of torsade de pointes. The QT interval in the sinus beat immediately preceding the torsade de pointes is 600 ms (reproduced with permission from Reference KhanKhan, 2002).
Repolarisation effects induced by antipsychotics, particularly thioridazine and chlorpromazine, have long been recognised in the form of T-wave changes on the ECG. For example, 58% of patients treated with 100–800 mg thioridazine daily showed some type of T-wave changes (Reference WendkosWendkos, 1967). These have been graded into three categories based on direction, amplitude and T-wave structure: grade 1 (T-wave broadening, blunting without loss of amplitude); grade 2 (loss of amplitude and bifid T-wave); grade 3 (flattened/inverted T-wave). All T-wave changes normalised in the majority of phenothiazine-treated patients after overnight fasting alone, and the remainder reverted to normal after oral potassium administration (Fig. 3). Oral administration of isosorbide dinitrate, potassium or ergot alkaloids also reversed thioridazine-induced T-wave changes, but was unsuccessful in reversing T-wave changes arising from cardiac pathology or quinidine therapy. Hence, it has been suggested that some antipsychotic repolarisation effects may be ‘benign’ (Reference WendkosWendkos, 1967) and that not all are necessarily associated with the more serious cardiac consequences reported in more recent literature (Reference Reilly, Ayis and JonesReilly et al, 2000). The impact of fasting, however, illustrates the effects of diet on ECG changes.

Fig. 3 ECG of a phenothiazine-treated patient: complete normalisation of repolarisation abnormalities by overnight fasting. Left panel: initial non-fasting T-wave changes; right panel: T-wave normalisation after overnight fasting (reproduced with permission from Reference Alvarez-Mena and FrankAlvarez-Mena & Frank, 1973).
The QT-prolonging tendency of antipsychotics, which is shared to a lesser extent by tricyclic antidepressants (Reference Reilly, Ayis and JonesReilly et al, 2000), is based on their capacity to block IKr, the delayed rectifier, the principal repolarising current terminating ventricular action potential in phase 2–3 (Fig. 4). Blockade of IKr (encoded by the HERG gene) may cause sufficient QT prolongation to generate extra potentials, known as early after-depolarisations, and to distort phase 3 of normal repolarisation (Figs 4 & 5). Early after-depolarisations may give rise to ventricular premature beats capable of triggering a run of torsade de pointes, especially if these beats distort the preceding T-wave (R-on-T) or conform to a long–short coupling pattern (Fig. 6). The risk of drug-induced torsade de pointes is increased under certain conditions (Boxes 2 & 3).

Fig. 4 Schematic representation of the action potential and corresponding ECG phases.
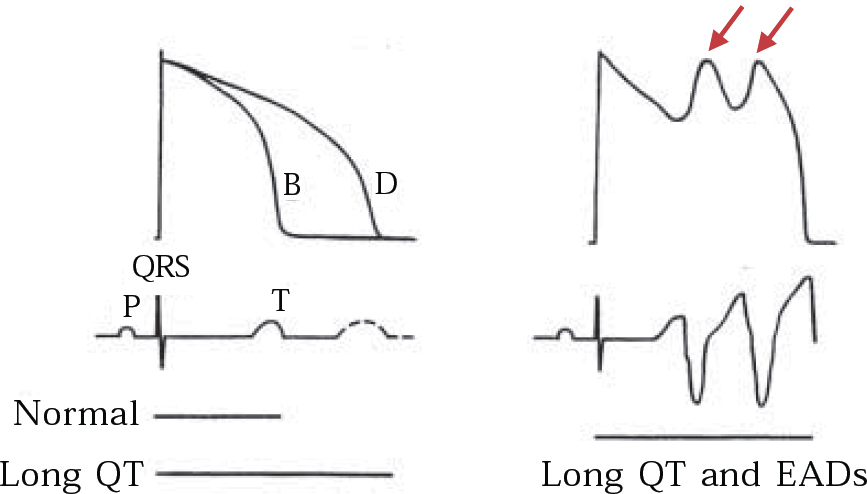
Fig. 5 Schematic representation of IKr block on the QT interval: (a) monophasic action potential (upper trace) and corresponding surface ECG (lower trace) before (B) and after (D) exposure to IKr blocking drug and the effect on the QT interval; (b) prolonged QT interval induces two early after-depolarisations (EADs) in the action potential (arrows), thus giving rise to a run of torsade de pointes on the surface ECG (lower trace). Reproduced with permission from Reference TamargoTamargo (2000).
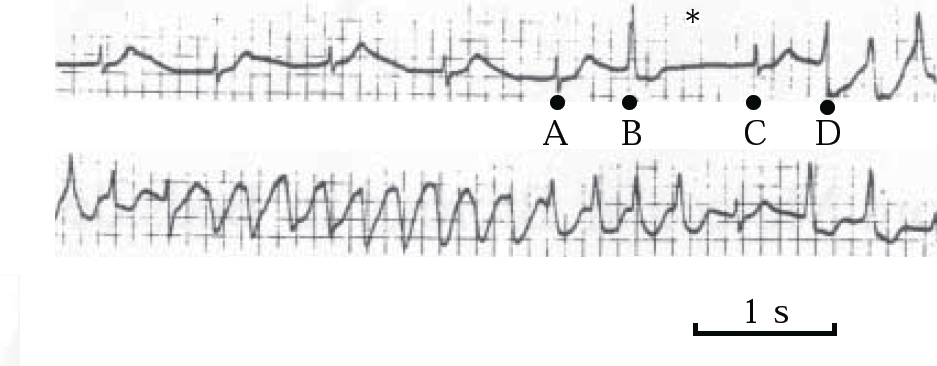
Fig. 6 ECG demonstrating long–short coupling sequence. A ventricular premature contraction (B) succeeds a sinus beat (A), leading to a compensatory pause (long cycle ∗); another sinus beat (C), followed by a second ventricular premature contraction (D) (short cycle), triggers a run of torsade de pointes. Reproduced with permission from Reference TamargoTamargo (2000).
Box 2 Drugs and conditions associated with acquired long QT syndromes
Antiarrhythmic drugs
Antipsychotics
Antidepressants
Antihistamines
Antimicrobials
Structural heart disease
Bradycardias
Intracranial lesions
Electrolyte abnormalities
Alcoholism
Anorexia nervosa
Hypothyroidism
Box 3 Risk of drug-induced torsade de pointes
Pre-existing QT prolongation
Pre-existing QT dispersion
Hypokalaemia
Hypomagnesaemia
Severe hypocalcaemia
QT-prolonging co-therapy
Sinus bradycardia <50 bpm
Pre-exisiting polymorphic ventricular premature beats showing R-on-T or long–short cycles
Normal QT values are not universally established because so many variables affect measurement. These include gender (longer in females), time of day (longer in sleep and early morning), diet, heart rate (QTc denotes QT corrected for heart rate using Bazett, Friderica or other regression formulae) and lead selection (longer in leads II, V2 and V3). However, a consensus appears to be emerging of a normal QTc upper limit of 450 ms for males and 470 ms for females, with a ‘red zone’ limit of 500 ms for both genders. Reference Bednar, Harrigan and AnzianoBednar et al (2001) warn that intervals in excess of these values should raise concern. In a literature review of 116 torsade de pointes cases associated with non-cardiac treatment, they found that 92% had QTc intervals >500 ms.
In terms of QTc change from baseline on treatment, it has likewise been recommended, notwithstanding a tendency for regression towards the mean, that an increase of 30 ms is a potential cause for concern and that a 60 ms increase is a definite cause for concern (Reference Bednar, Harrigan and AnzianoBednar et al, 2001). In addition, a QT dispersion of 40–60 ms is considered acceptable, but it is thought to be pathological when it exceeds 100 ms or is 100% increased from baseline values (the QT dispersion is the difference between the longest and shortest QT on a 12-lead ECG, which is widely although not universally regarded as reflecting repolarisation asynchrony across the entire ventricle and another risk factor for torsade de pointes) (Reference Bednar, Harrigan and AnzianoBednar et al, 2001).
Antipsychotics differ in their capacity for QT prolongation. In vitro studies using HERG-transfected cell lines demonstrate a concentration-dependent effect for IKr blocking (Table 1). This is consistent with findings from retrospective systematic clinical studies identifying thioridazine and the tricyclic antidepressants as predictive of QT prolongation (Reference Reilly, Ayis and JonesReilly et al, 2000).
Table 1 Drug concentrations for blocking HERG tail-currents (IKr) in HERG-transfected cell lines

| Drug | Potency (in descending order) |
|---|---|
| Sertindole1 | 2.9 nM |
| Haloperidol1 | 1.0 μM |
| Thioridazine2 | 1.07± 0.06 μM |
| Chlorpromazine2 | 1.47±0.03 μM |
| Clozapine2 | 2.63±0.12 μM |
| Amitriptyline2 | 10.0±1.1 μM |
The prevalence of torsade de pointes in the psychiatric population is unknown, but estimates from antiarrhythmic-induced torsade de pointes in the cardiac population ranges from 3% to 15%. Being associated with entirely non-specific symptoms such as palpitations, dizziness, syncope and seizures, its potential seriousness may be easily misconstrued as primary psychiatric disorder: this can have fatal outcome. Although usually self-limiting, torsade de pointes tends to recur and in 31% of cases progresses to ventricular fibrillation and sudden death.
These QT-prolonging proarrhythmic concerns led to the placing of prescribing restrictions on thioridazine, a voluntary withdrawal of droperidol in the UK by its manufacturer and a complete withdrawal of sertindole from the European market. They have also shifted the cardiac focus onto other increasingly prescribed atypical antipsychotics. For example, after some initial concern the US Food and Drugs Administration requested Study 054 (Psychopharmacological Drugs Advisory Committee, 2000) to further clarify the QT-prolonging capacity of ziprasidone and to compare it with olanzapine, risperidone, quetiapine, thioridazine and haloperidol in an open-label randomised 4-week trial involving 183 patients with psychosis. The mean QTc change from baseline was 10 ms greater with ziprasidone than with olanzapine, risperidone and quetiapine, and 10 ms less than with thioridazine; haloperidol demonstrated the least change and all agents showed a dose-dependent effect. Ziprasidone was subsequently licensed for use in the USA, and although a black-box warning was avoided, a bold-type warning of its QT-prolonging capacity was included.
Additional risks factors for QT-prolongation and torsade de pointes in the psychiatric population include deliberate or accidental antipsychotic overdose, effects of concomitant QT-prolonging medication (either indirect such as CYP 450 3A3 and 3A4 inhibitors, fluoxetine, fluvoxamine, grapefruit juice and ketoconazole, or direct such as tricyclic antidepressants, antihistamines and anti-infectives), comorbid substance misuse and, in particular, the effects of high sympathetic arousal during restraint. Such arousal releases large amounts of circulating catecholamines which, in addition to increasing cardiac oxygen consumption, lower serum levels by β-2 adrenergic stimulation of potassium (K+) uptake into skeletal muscle cells. Reference Hatta, Takahashi and NakamuraHatta et al (1999) found a significant negative correlation between serum K+ and high v. low levels of psychotic agitation in acute in-patients (3.59 v. 3.79 mEq/l), and lower serum K+ in more agitated acute in-patients compared with out-patients. Thus, in a control and restraint situation, the QT-prolonging effects of hypokalaemia and intramuscular antipsychotics, together with the potential for hypoxia and myocardial irritability from inadvertent ventilatory embarrassment and greater cardiac strain, may result in the kind of potent proarrhythmic cocktail that prompted the Collge to warn that ‘the risk of sudden cardiotoxic collapse in response to antipsychotic medication given during a period of high physiological arousal should be widely publicised’(Royal College of Psychiatrists, 1997).
Coronary artery disease
Major and minor depression have an important relationship with coronary artery disease. Many studies have demonstrated a strong association between depression and coronary artery disease: there is increased incidence of the disease in patients with depression who have an otherwise healthy heart, and exacerbation of existing coronary artery disease and cardiac mortality in the 15–20% of post-infarction patients who develop major, and even minor, depression. Even sub-syndromal depressive symptoms therefore increase cardiac risk in coronary artery disease (Reference Penninx, Beekman and HonigPenninx et al, 2001). This strong association persists even after controlling for established cardiac risk factors such as smoking and diabetes.
Panic disorder and phobic anxiety states are also associated with increased rates of coronary artery disease and cardiac mortality, possibly mediated by coronary artery spasm and sympathetic-mediated cardiac arrhythmias, but the association is less well studied and documented (Reference Mansour, Wilkinson and JenningsMansour et al, 1998). Serotonin (5-HT) activity represents one possible link between depression, anxiety and coronary artery disease. Activated platelets, apart from platelet factor-4 (PF-4) and b-thromboglobulin (b-TG), release large quantities of serotonin. This has two main effects: first, it causes vasodilation in normal, but vasoconstriction in diseased, coronary vessels; and second, it promotes endothelial vessel dysfunction, with local further platelet activation and endothelial smooth-muscle proliferation, potentially responsible for local atherogenesis.
Levels of PF-4 and b-TG can be assayed, and are higher in patients with coronary artery disease who have depression than in similar patients without depression and in healthy controls (Reference Laghrissi-Thode, Wagner and PollockLaghrissi-Thode et al, 1997).
Because increased platelet activation may thus contribute to atherogenesis, coronary artery disease and sudden death, attempts to reduce platelet activity in 'coronary-prone’ individuals with depression or anxiety may become an important aspect of cardiac risk reduction, especially since aspirin therapy alone does not appear to reduce platelet activity in this population (Reference Laghrissi-Thode, Wagner and PollockLaghrissi-Thode et al, 1997).
Paroxetine appears to be a promising agent in this regard. Reference Musselman, Marzec and ManatungaMusselman et al (2000) report a study of 15 patients with depression who showed no evidence of coronary artery disease, although some did have known risk factors for the disease. At the start of the study, all had elevated PF-4 levels. After receiving 20 mg paroxetine daily for 6 weeks, a significant reduction in these levels was observed in both those with and without known risk factors for coronary artery disease. Roose and colleagues found further evidence supporting the use of paroxetine in co-morbid depression or anxiety and coronary artery disease. They compared paroxetine 20–30 mg daily with therapeutic doses of nortriptyline in 81 patients with coronary artery disease and depression. Despite equal antidepressant efficacy after 6 weeks’ treatment, paroxetine had more benefit on cardiac function, including heart rate, heart rate variability, rhythm and adverse cardiac events (Reference Roose, Laghrissi-Thode and KennedyRoose et al, 1998b ).
The best evidence to date for antidepressant intervention in this patient group has come from the Sertraline Antidepressant Heart Attack Randomised Trial (SADHART; Reference Glassman, O'Connor and CaliffGlassman et al, 2002). A study of 369 patients recently discharged following acute myocardial infarction or unstable angina, who were randomised to sertraline 50–200 mg/day or to placebo, demonstrated treatment safety and efficacy of the antidepressant over placebo.
The safety and benefits of SSRIs, especially paroxetine and fluoxetine, over tricyclic antidepressants on general cardiac function, platelet coagulability and, possibly, atherogenesis appears to recommend their use as psychotropics of first choice in patients with, or at high risk of, coronary artery disease, especially smokers, postmenopausal women and patients with hypertension, diabetes, hyperlipidaemia and/or a strong family history of coronary artery disease.
Contractility and effects on cardiac muscle
Radionuclide ventriculography has demonstrated, contrary to original concerns, that psychotropic drugs do not exert a direct negative inotropic effect (Reference Roose, Glassman and AttiaRoose et al, 1998a ). However, antipsychotics (clozapine in particular) are occasionally associated with an increased risk of myocarditis or cardiomyopathy (Reference Kilian, Kerr and LawrenceKilian et al, 1999). Kilian and colleagues reported 15 cases of myocarditis and 8 of dilated cardiomyopathy at normal daily doses (100–725 mg clozapine) in 8000 Australian patients. All but 3 were male, with a mean age of 36. The myocarditis occurred in the first month of treatment (often from hypersensitive eosinophilic myocarditis), had few specific symptoms and frequently presented with sudden death. Cardiomyopathy developed more slowly, within the first 12 months, usually presented with cough, dyspnoea, palpitations or heart failure, could improve on clozapine discontinuation and appeared to have a better prognosis. These and other reports have led to increased warnings of these cardiac complications in information on prescribing and of the need for early investigation and clozapine cessation in suspect cases.
As eosinophilic myocarditis seems to be the favoured aetiology, blood eosinophilia should be carefully sought. However, a raised plasma level of eosinophil cationic protein, an assayable pro-inflammatory protein released from degranulated eosinophils, seems a preferable marker as it remains elevated even in cases with normal eosinophil counts (Reference Arima, Kanoh and KawanoArima et al, 2002).
Blood pressure
Postural (orthostatic) hypotension has many causes but is probably the most common and potentially most troublesome psychotropic side-effect (Box 4). Usually defined as a ≥20 mmHg reduction in systolic blood pressure after 1–3 minutes of standing upright, it may cause symptomatic underperfusion of vital organs, leading to dizziness, blurred vision, fainting, falls, fractures, seizures, strokes and cardiac ischaemia, especially in elderly people. Fainting usually occurs when mean arterial pressure (diastolic plus 1/3 pulse pressure) drops to 70 mmHg or below, corresponding to an upper-arm blood pressure of 90/60 or less.
Box 4 Causes of postural hypotension (after Reference Engstrom and AminoffEngstrom & Aminoff,1997, with permission)
Cardiac pump failure
Myocardial infarction
Myocarditis
Constrictive pericarditis
Aortic stenosis
Tachy- or bradyarrhythmias
Venous pooling
Alcohol
Postprandial dilatation of splanchnic vessel beds
Vigorous exercise with dilatation of skeletal vessel beds
Hot baths/showers/fever/weather
Prolonged recumbency or standing
Reduced intravascular volume
Straining-toilet/coughing/lifting
Dehydration/vomiting/diarrhoea
Haemorrhage/burns
Salt-losing nephropathy
Adrenal insufficiency
Diabetes insipidus
Other causes
Shy–Drager syndrome
Diabetic neuropathy
Alcoholic neuropathy
Porphyria
Anorexia nervosa
Riley–Day syndrome
Chronic fatigue syndrome
Medications
Antihypertensives
Diuretics/disulfiram
Vasodilators
Alpha- and beta-blockers
Barbiturates/opiates
Neuroleptics/tricyclics/MAOIs
Clinical studies of tricyclic antidepressants focusing mainly on imipramine and nortriptyline found a 20% prevalence of postural hypotension in treated patients. Non-standardised methodology may account for some conflicting findings, but the data suggest, first, a greater tendency to postural hypotension for tertiary tricyclics (e.g. imipramine and amitriptyline) than for secondary (e.g. nor-triptyline); second, that the degree of pretreatment postural blood pressure drop predicts on-treatment symptoms and when this pretreatment orthostatic hypotension was controlled for, removed age by itself as a predictor of postural drop; and third, that (counterintuitively) dose reduction may not relieve symptoms (Reference Glassman, Bigger and GiardinaGlassman et al, 1979).
There are fewer studies of MAOIs, for which the hypotensive mechanism remains elusive. In patients with depression, phenelzine 60 mg daily induced both a fall in supine systolic blood pressure and a postural drop. In 14% of patients, the latter caused significant symptoms, which reached a maximum after 4 weeks’ treatment. This contrasts with findings for imipramine, where supine blood pressure remained unaffected but the postural effect tended to maximise after 1 week. However, these results might be related to an early rapid dose escalation. Isocarboxazid and tranylcypromine are likewise associated with an 11% and 17% prevalence of symptomatic postural hypotension respectively (Reference Warrington, Padgham and LaderWarrington et al, 1989).
Postural hypotension is a well-documented effect of typical antipsychotics, but few systematic studies exist. In a study of 196 chronic in-patients with schizophrenia on stable antipsychotic treatment, symptomatic postural hypotension occurred in 77% within 1 minute, reducing to 17% after 3 minutes, on standing from supine. No other correlation with age, dose or potency of α1-adrenergic blocking drug emerged, although thioridazine was associated with greater postural blood pressure drop (Reference Silver, Kogan and ZlotogorskiSilver et al, 1990).
Atypical antipsychotics pose much less of a problem (with the possible exception of clozapine, where α1-adrenergic blockade is generally assumed to be responsible; it tends to occur early in treatment, particularly with rapid dose escalation, but tolerance usually builds up).
Care is needed when co-prescribing psychotropics and antihypertensives. Antipsychotics may, like tricyclics, antagonise centrally acting drugs such as clonidine and methyldopa. These are infrequently used as antihypertensives, but clonidine is popular in psychiatric practice for acute opiate detoxification. However, co-prescribing antipsychotics, especially low-potency typicals such as chlorpro-mazine or the atypicals clozapine or quetiapine, with any of the following may provoke profound postural hypotension: α1-adrenergic antagonists (e.g. doxazosin, which is also a treatment for benign prostatic hypertrophy); calcium-channel blockers (e.g. nifedipine); angiotensin-converting enzyme inhibitors (captopril); angiotensin-II receptor antagonists (losartan); or vasodilators (e.g. nitro-prusside). Postural hypotension is also a risk when antipsychotics are taken with β-blockers (probably because of pharmacokinetic interaction) or with diuretics (because of Na+ or volume depletion). The same hypotensive effects might be anticipated when tricyclic antidepressants or MAOIs are co-prescribed with peripheral antihypertensive agonists. One possible exception concerns phenelzine, whose hypotensive action was reversed on co-therapy with atenolol (Reference Merikangas and MerikangasMerikangas & Merikangas, 1995).
Symptomatic postural hypotension is associated with dizziness, blurred vision, syncope, falls and injuries, seizures and myocardial infarction. Effective management should therefore address prevention as well as specific therapy. Patient counselling is the most important aspect of prevention (Box 5). Extra vigilance in older people and patients who are dehydrated or have diabetes or hypertension on treatment is essential.
Box 5 Prevention of postural hypotension
Avoid prolonged standing
Adopt slow, careful posture changes when rising from bed or chair after prolonged rest
Ascend and descend stairs on your bottom
Sit, do not stand, in hot water
Be careful getting out of a hot bath
Be careful in hot environments
Make a tight fist before standing (isometric handgrip exercises have a pressor effect)
Drink cold and caffeinated drinks
Ensure copious fluid intake
Ensure adequate salt intake
Eat frequent but small meals
Avoid alcohol
Avoid vigorous exercise
Wear support stockings
Sleep with the bed head raised by 15–30 cm (a 30° tilt minimises nocturnal diuresis)
In addition to the simple measures shown in Box 5, fludrocortisone therapy might also be necessary. This acts by increasing arteriolar sensitivity to noradrenalin and, in bigger doses, by expanding plasma volume. Doses begin at 50 μg daily, increased by 50 μg increments to a maximum of 300 μg, according to response and level of hypotensive symptoms. The drug can usually be withdrawn after the early weeks of psychotropic treatment, as accomodation to the hypotensive effect usually develops. Fludrocortisone side-effects include fluid overload and heart failure, hypokalaemia and supine hypertension.
Conclusion
On the basis of the literature referenced in this paper, it could be argued that all mental health units have ready access to ECG monitoring equipment. Modern machines provide an automated printout that can be of great assistance to the psychiatrist in ECG interpretation, but in cases of doubtful validity, cardiology colleagues are usually most helpful in providing manual interpretation. Clinicians with a particular interest in this field may wish to conduct ECG studies in a greater proportion of their patients or pursue more complex studies involving heart rate variability, which would probably involve collaboration with cardiology services anyway .
Patients already at higher risk of cardiac complications will continue to be closely monitored. The difficulties of widespread monitoring of the psychiatric population, apart from practical considerations, would appear too considerable to justify at present, and cardiologists or other psychiatrists with particular knowledge of the field have not thus far endorsed its introduction.
Monitoring of pulse and blood pressure ought to remain essential components of the psychiatric examination. It could also be argued that psychiatric in-patients perceived to be at greater risk for QTc prolongation might receive more careful monitoring to minimise known risks (especially low serum K+ and Mg2+) and have their levels maintained well into the normal range (serum K+ > 3.8 mmol/l) by a diet rich in potassium and magnesium or, if necessary, by oral supplementation.
Furthermore, control and restraint procedures for disturbed patients must be careful to avoid any restriction of chest or diaphragmatic ventilation, which might add to any pre-existing cardiac risk. If rapid tranquillisation becomes necessary, and if time and circumstances allow, intramuscular benzo-diazepines (which have negligible cardiotropic effects) could be administered initially, to be followed with an intramuscular antipsychotic given soon after, when the patient has begun to settle.
Finally, as there is a possible association with myocarditis and cardiomyopathy, any patients commencing on clozapine could be considered for pre-treatment baseline ECG, chest X-ray, eosinophil counts and eosinophil cationic protein level, repeated as necessary. Subsequent comparison with baseline would help appropriate interpretation of treatment-emergent changes.
Multiple choice questions
-
1 Torsade de pointes:
-
a is a variety of French perfume
-
b is a polymorphic ventricular tachycardia
-
c is associated with sudden death
-
d is more likely to occur with hypokalaemia
-
e is associated with long–short coupling on the ECG.
-
-
2 A prolonged QT interval:
-
a is associated with torsade de pointes
-
b may be congenital or acquired
-
c is associated with antipsychotic medication
-
d is more common in women
-
e is associated with HERG/IKR blockade.
-
-
3 Myocardial infarction:
-
a has a higher incidence in patients with depression who have no cardiac disease
-
b may present with postural hypotension
-
c may be associated with prolonged QT interval
-
d is associated with increased platelet reactivity
-
e is associated with sudden death when complicated by reduced heart rate variability.
-
-
4 Tricyclic antidepressants:
-
a are type 1 antiarrhythmic agents
-
b block sodium and potassium cardiac channels
-
c are contraindicated in post-myocardial infarction depression
-
d decrease heart rate variability
-
e are a safe treatment for depression in the presence of bundle branch block.
-
-
5 Postural hypotension:
-
a is a rare complication of psychotropic drug treatment
-
b may be a manifestation of myocarditis
-
c may present with falls, fits or faints
-
d is usually detectable on clinical examination
-
e is more frequent with SSRI than with tricyclic antidepressant therapy.
-
MCQ answers

| 1 | 2 | 3 | 4 | 5 | |||||
| a | F | a | T | a | T | a | T | a | F |
| b | T | b | T | b | T | b | T | b | T |
| c | T | c | T | c | T | c | T | c | T |
| d | T | d | T | d | T | d | T | d | T |
| e | T | e | T | e | T | e | F | e | F |








eLetters
No eLetters have been published for this article.